量化理论计算探究薁(azulene)的反Kasha规则荧光机制
根据Kasha规则,由于高激发态之间存在快速的无辐射跃迁,分子的荧光或磷光的初始状态是最低的单重态或三重态。薁(azulene)是典型违反 Kasha 规则的例子。azulene的荧光来自于S2态,可以认为是由于S2→S1能隙比较大,所以降低了S2→S1内转换速率。另外,由于S1和S0之间的能隙相对较小,所以其内转换速率很大,从而降低了S1→S0的荧光量子效率,所以S1→S0的荧光很难被发现。这里以具有反常荧光现象的azulene为例,使用BDF软件和MOMAP软件,计算azulene的S1→S0的辐射速率和内转换速率,从而解释azulene第一激发态极低的量子效率导致其荧光难以被观测到的实验结果。
MOMAP对azulene的S1→S0的辐射速率和内转换速率的计算需要BDF量化软件的结构优化频率结果文件、非绝热耦合结果文件和参数部分都已完成。首先完成量化软件BDF的计算部分。
BDF计算部分
准备azulene分子结构的xyz文件如下:
18
C -0.48100000 0.74480000 0.00000000
C -0.56240000 -0.71320000 0.00020000
C -1.75790000 1.17860000 -0.00030000
C -1.96510000 -1.08880000 0.00000000
C 0.66870000 1.60890000 0.00030000
C 0.45100000 -1.58850000 0.00030000
C -2.66930000 0.05180000 -0.00010000
C 1.95140000 1.22210000 0.00020000
C 1.86730000 -1.29960000 -0.00020000
C 2.49720000 -0.11610000 -0.00040000
H -2.09080000 2.20560000 -0.00040000
H -2.35750000 -2.09240000 0.00010000
H 0.46620000 2.67860000 0.00070000
H 0.22090000 -2.65340000 0.00040000
H -3.74370000 0.14050000 -0.00030000
H 2.70720000 2.00650000 0.00050000
H 2.49320000 -2.19150000 -0.00060000
H 3.58670000 -0.13930000 -0.00080000
打开Device studio,点击File-new project,命名为 fluorescence.hpf ,将 azulene.xyz 拖入Project中,双击 azulene.hzw ,得到如图所示界面。

首先使用BDF进行azulene的结构优化和频率计算。选中Simulator → BDF → BDF,界面中设置参数。在Basic Settings界面中的Calculation Type选择Opt+Freq,方法采用默认的GB3LYP泛函,基组在Basis中的All Electron类型中,选择6-31G(d,p)。Basic Settings界面中的其它参数以及SCF Settings、OPT Settings、Freq Settings等面板的参数使用推荐的默认值,不需要做修改。之后点击 Generate files 即可生成对应计算的输入文件。
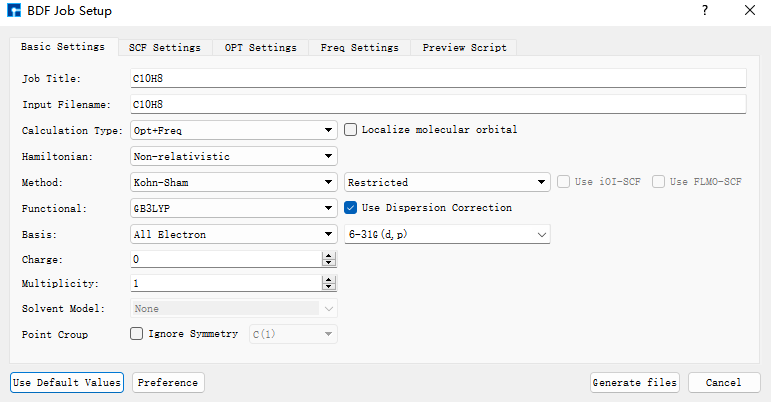
选中生成的 bdf.inp 文件,右击选择open with,打开该文件,如下所示:
$compass
Title
C10H8
Geometry
C -0.48100000 0.74480000 0.00000000
C -0.56240000 -0.71320000 0.00020000
C -1.75790000 1.17860000 -0.00030000
C -1.96510000 -1.08880000 0.00000000
C 0.66870000 1.60890000 0.00030000
C 0.45100000 -1.58850000 0.00030000
C -2.66930000 0.05180000 -0.00010000
C 1.95140000 1.22210000 0.00020000
C 1.86730000 -1.29960000 -0.00020000
C 2.49720000 -0.11610000 -0.00040000
H -2.09080000 2.20560000 -0.00040000
H -2.35750000 -2.09240000 0.00010000
H 0.46620000 2.67860000 0.00070000
H 0.22090000 -2.65340000 0.00040000
H -3.74370000 0.14050000 -0.00030000
H 2.70720000 2.00650000 0.00050000
H 2.49320000 -2.19150000 -0.00060000
H 3.58670000 -0.13930000 -0.00080000
End Geometry
Basis
6-31G(d,p)
Skeleton
Group
C(1)
$end
$bdfopt
Solver
1
MaxCycle
108
IOpt
3
Hess
final
$end
$xuanyuan
Direct
$end
$scf
RKS
Charge
0
SpinMulti
1
DFT
GB3LYP
D3
MPEC+COSX
Molden
$end
$resp
Geom
$end
选中 bdf.inp 文件,右击选择Run,弹出如下界面:
点击Run提交作业,会自动弹出结果文件的实时输出。

任务结束后 bdf.out , bdf.out.tmp , bdf.scf.molden 三个结果文件会出现在Project中。

选择 bdf.out ,右击show view,在Optimization对话框中,显示结构已经达到收敛标准。

在Frequency对话框中,检查频率,若不存在虚频证明结构已经优化到极小点。
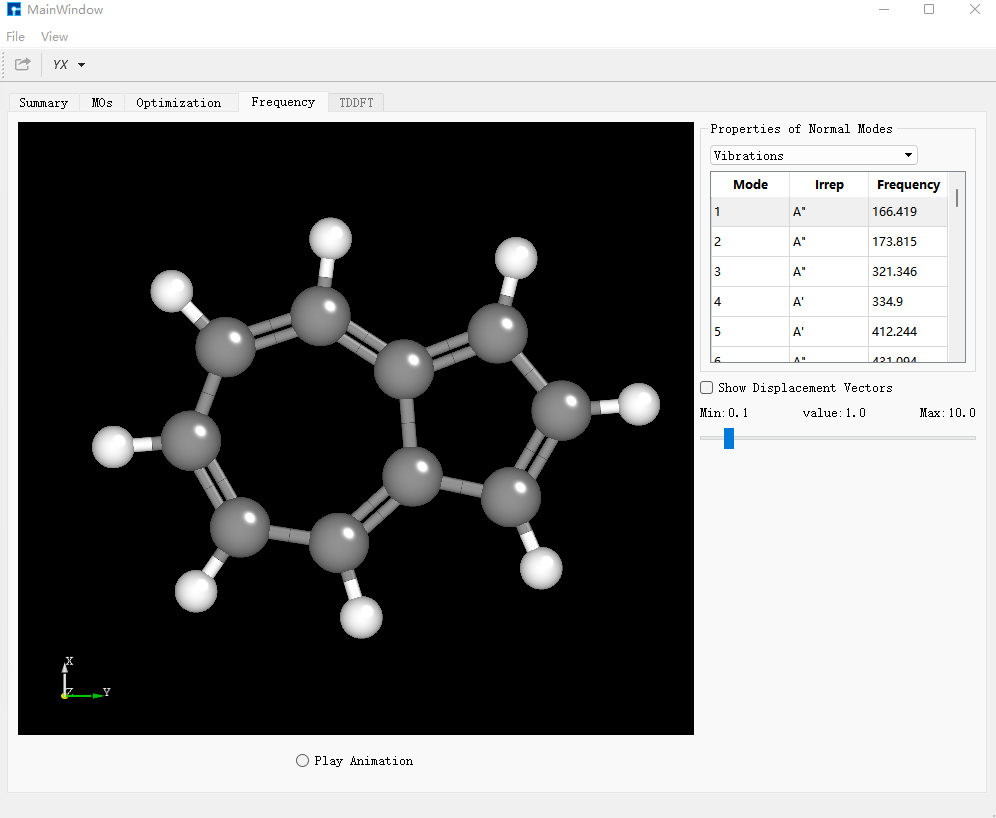
在Summary对话框中,Total Energy为-385.87807600 a.u.,为需要的基态azulene的单点能。

点击Job Manager中该计算任务,点击服务器,已经进入到了该任务所在文件夹下,输入 /data/hzwtech/DS-BDF_2022A/sbin/optgeom2xyz.py bdf.optgeom ,回车,生成 bdf.xyz 文件。点击文件传输工具,进入文件夹下,将 bdf.xyz 文件拖出,即为下一步激发态结构优化需要的输入文件。改名为 azulene_s0.xyz ,打开文件夹,将第二行描述行去掉,拖入Device Studio中。
使用BDF进行azulene的S1激发态结构优化和频率计算。选中Simulator → BDF → BDF,界面中设置参数。在Basic Settings界面中的Calculation Type选择TDDFT-OPT+Freq,方法采用默认的GB3LYP泛函,基组在Basis中的All Electron类型中,选择6-31G(d,p)。SCF Settings和TDDFT Settings面板中将Use MPEC+COSX Acceleraton的默认勾选去掉。Basic Settings、SCF Settings、TDDFT Settings界面中的其它参数以及OPT Settings、Freq Settings等面板的参数使用推荐的默认值,不需要做修改。之后点击 Generate files 即可生成对应计算的输入文件。
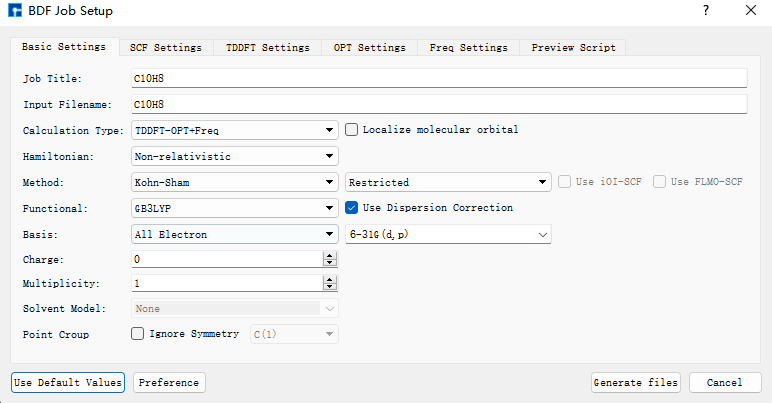


选中生成的 bdf.inp 文件,右击选择open with,打开该文件,如下所示:
$compass
Title
C10H8
Geometry
C 0.79273796 0.49102542 -0.00003307
C -0.70229649 0.61186591 0.00000000
C 1.30022932 1.80163337 -0.00006272
C -0.99262499 1.98726800 0.00007812
C 1.54415132 -0.67887418 0.00000000
C -1.63318173 -0.42094563 -0.00002837
C 0.21877157 2.69859813 0.00000000
C 1.10656346 -2.00562676 0.00005788
C -1.41619168 -1.80093044 -0.00004814
C -0.20258112 -2.49333483 0.00000000
H 2.35092512 2.06249889 -0.00009828
H -1.98777600 2.41348149 0.00017650
H 2.62424717 -0.53731745 0.00001117
H -2.67585843 -0.10561277 -0.00001521
H 0.30641472 3.77916915 0.00002386
H 1.88966566 -2.75951313 0.00017581
H -2.31053950 -2.41870505 -0.00009019
H -0.29054446 -3.57807510 0.00000000
End Geometry
Basis
6-31G(d,p)
Skeleton
Group
C(1)
$end
$bdfopt
Solver
1
MaxCycle
108
IOpt
3
Hess
final
$end
$xuanyuan
Direct
$end
$scf
RKS
Charge
0
SpinMulti
1
DFT
GB3LYP
D3
Molden
$end
$tddft
Imethod
1
Isf
0
Idiag
1
Iroot
6
Istore
1
$end
$resp
Geom
Method
2
Nfiles
1
Iroot
1
$end
选中 bdf.inp 文件,右击选择Run提交作业,任务结束后 bdf.out , bdf.out.tmp , bdf.scf.molden 三个结果文件会出现在Project中。
选择 bdf.out ,右击show view,在Optimization对话框中,显示结构已经达到收敛标准。

在Frequency对话框中,检查频率,若不存在虚频证明结构已经优化到极小点。

选择 bdf.out.tmp ,右击open with containing folder,打开 bdf.out.tmp ,在文件开始向下查找第一个tddft计算模块 module tddft 。根据tddft模块的 Ground to excited state Transition electric dipole moments (Au) 中的State 1的跃迁电偶极矩,得到momap需要的参数EDMA,计算方法为: \(\sqrt{(0.3456)^2+(-0.1159)^2+(-0.0000)^2}\) = 0.3646 a.u.。需要将单位a.u.转换为Debye,因此EDMA= 0.3646*2.5417=0.9267 Debye。
*** Ground to excited state Transition electric dipole moments (Au) ***
State X Y Z Osc.
1 0.3456 -0.1159 0.0000 0.0079 0.0079
2 0.0538 0.1576 0.0000 0.0025 0.0025
3 -0.6407 0.2166 0.0000 0.0527 0.0527
4 0.9123 2.7142 0.0000 1.0366 1.0366
5 -0.0001 0.0002 0.0200 0.0001 0.0001
6 -1.2021 0.4024 -0.0000 0.2383 0.2383
在文件末尾向上查找第一个tddft计算模块 module tddft 。根据tddft模块的 Ground to excited state Transition electric dipole moments (Au) 中的State 1的跃迁电偶极矩,得到momap需要的参数EDME,计算方法为: \(\sqrt{(-0.2427)^2+(0.0816)^2+(-0.0000)^2}\) = 0.2560 a.u.。需要将单位a.u.转换为Debye,因此EDMA= 0.3646*2.5417=0.6507 Debye。
*** Ground to excited state Transition electric dipole moments (Au) ***
State X Y Z Osc.
1 -0.2427 0.0816 0.0000 0.0026 0.0026
2 0.0403 0.1199 0.0000 0.0013 0.0013
3 -0.2655 0.0888 -0.0000 0.0090 0.0090
4 -0.8679 -2.5831 0.0000 0.8696 0.8696
5 -1.2356 0.4150 0.0000 0.2404 0.2404
6 -0.0008 0.0003 0.0006 0.0000 0.0000
在该 tddft 模块的 Statistics for [dvdson_rpa_block]: 中读取No. 1态的能量为 -385.8030480568 a.u.,即为S1态的单点能。
------------------------------------------------------------------
Statistics for [dvdson_rpa_block]:
No. of blocks = 1
Size of blocks = 50
No. of eigens = 6
No. of HxProd = 73 Averaged = 12.167
Eigenvalues (a.u.) =
0.0593694732 0.1241240214 0.1718082072
0.1756634611 0.2122947029 0.2131964457
------------------------------------------------------------------
No. 1 w= 1.6155 eV -385.8030480568 a.u. f= 0.0026 D<Pab>= 0.0000 Ova= 0.4881
CV(0): A( 33 )-> A( 36 ) c_i: -0.1361 Per: 1.9% IPA: 5.454 eV Oai: 0.7151
CV(0): A( 34 )-> A( 35 ) c_i: -0.9847 Per: 97.0% IPA: 2.582 eV Oai: 0.4815
No. 2 w= 3.3776 eV -385.7382935086 a.u. f= 0.0013 D<Pab>= 0.0000 Ova= 0.8097
CV(0): A( 33 )-> A( 35 ) c_i: -0.6839 Per: 46.8% IPA: 4.083 eV Oai: 0.8197
CV(0): A( 34 )-> A( 36 ) c_i: 0.7092 Per: 50.3% IPA: 3.953 eV Oai: 0.8078
No. 3 w= 4.6751 eV -385.6906093228 a.u. f= 0.0090 D<Pab>= 0.0000 Ova= 0.7195
CV(0): A( 32 )-> A( 35 ) c_i: 0.5413 Per: 29.3% IPA: 5.770 eV Oai: 0.7332
CV(0): A( 33 )-> A( 36 ) c_i: 0.8170 Per: 66.7% IPA: 5.454 eV Oai: 0.7151
CV(0): A( 34 )-> A( 38 ) c_i: 0.1417 Per: 2.0% IPA: 7.164 eV Oai: 0.7494
No. 4 w= 4.7800 eV -385.6867540689 a.u. f= 0.8696 D<Pab>= 0.0000 Ova= 0.7950
CV(0): A( 32 )-> A( 36 ) c_i: 0.1707 Per: 2.9% IPA: 7.141 eV Oai: 0.8644
CV(0): A( 33 )-> A( 35 ) c_i: -0.6794 Per: 46.2% IPA: 4.083 eV Oai: 0.8197
CV(0): A( 33 )-> A( 38 ) c_i: 0.1022 Per: 1.0% IPA: 8.665 eV Oai: 0.8000
CV(0): A( 34 )-> A( 36 ) c_i: -0.6312 Per: 39.8% IPA: 3.953 eV Oai: 0.8078
No. 5 w= 5.7768 eV -385.6501228271 a.u. f= 0.2404 D<Pab>= 0.0000 Ova= 0.7166
CV(0): A( 31 )-> A( 36 ) c_i: -0.1797 Per: 3.2% IPA: 8.008 eV Oai: 0.7623
CV(0): A( 32 )-> A( 35 ) c_i: -0.7750 Per: 60.1% IPA: 5.770 eV Oai: 0.7332
CV(0): A( 33 )-> A( 36 ) c_i: 0.4518 Per: 20.4% IPA: 5.454 eV Oai: 0.7151
CV(0): A( 34 )-> A( 38 ) c_i: 0.1740 Per: 3.0% IPA: 7.164 eV Oai: 0.7494
CV(0): A( 34 )-> A( 40 ) c_i: -0.2680 Per: 7.2% IPA: 8.035 eV Oai: 0.6274
No. 6 w= 5.8014 eV -385.6492210843 a.u. f= 0.0000 D<Pab>= 0.0000 Ova= 0.4336
CV(0): A( 29 )-> A( 35 ) c_i: 0.9969 Per: 99.4% IPA: 7.064 eV Oai: 0.4335
将S1态的单点能与S0态的单点能相减,即得momap需要的两态的能量差参数Ead=0.07502 a.u.。
基于基态结构,做S0和S1之间的非绝热耦合计算。点击 azulene_s0.hzw ,右击点击copy,设置new file name为nacme,在Project中出现 nacme.hzw 。双击 nacme.hzw ,使用BDF进行azulene的非绝热耦合计算。选中Simulator → BDF → BDF,界面中设置参数。在Basic Settings界面中的Calculation Type选择TDDFT-NAC,
方法采用默认的GB3LYP泛函,基组在Basis中的All Electron类型中,选择6-31G(d,p)。SCF Settings和TDDFT Settings面板中将Use MPEC+COSX Acceleraton的默认勾选去掉。在TDDFT Settings面板中的Non-Adiabatic Coupling内容框中,在默认的Coupling between Ground and Excited-State下,点击”+”号,Basic Settings、
SCF Settings、TDDFT Settings界面中的其它参数以及OPT Settings、Freq Settings等面板的参数使用推荐的默认值,不需要做修改。之后点击 Generate files 即可生成对应计算的输入文件。

选中生成的 bdf.inp 文件,右击选择open with,打开该文件,如下所示:
$compass
Title
C10H8
Geometry
C 0.79273796 0.49102542 -0.00003306
C -0.70229648 0.61186591 0.00000000
C 1.30022931 1.80163336 -0.00006271
C -0.99262499 1.98726799 0.00007812
C 1.54415131 -0.67887417 0.00000000
C -1.63318173 -0.42094562 -0.00002837
C 0.21877157 2.69859812 0.00000000
C 1.10656346 -2.00562675 0.00005788
C -1.41619168 -1.80093044 -0.00004813
C -0.20258112 -2.49333482 0.00000000
H 2.35092512 2.06249888 -0.00009827
H -1.98777599 2.41348149 0.00017649
H 2.62424717 -0.53731744 0.00001117
H -2.67585843 -0.10561277 -0.00001520
H 0.30641472 3.77916915 0.00002385
H 1.88966565 -2.75951312 0.00017580
H -2.31053950 -2.41870504 -0.00009018
H -0.29054446 -3.57807510 0.00000000
End Geometry
Basis
6-31G(d,p)
Skeleton
Group
C(1)
$end
$xuanyuan
Direct
$end
$scf
RKS
Charge
0
SpinMulti
1
DFT
GB3LYP
D3
MPEC+COSX
Molden
$end
$tddft
Imethod
1
Isf
0
Idiag
1
Iroot
6
Istore
1
$end
$resp
Quad
FNAC
Norder
1
Method
2
Nfiles
1
Single
States
1
1 1 1
$end
选中 bdf.inp 文件,右击选择Run提交作业,任务结束后 bdf.out , bdf.scf.molden 三个结果文件会出现在Project中。
至此,MOMAP对azulene的S1→S0的辐射速率和内转换速率的计算需要的BDF量化软件的结构优化频率结果文件、非绝热耦合结果文件和参数部分都已完成。
MOMAP计算部分
在使用量化软件BDF完成azulene的基态和激发态的结构优化、频率计算以及非绝热耦合的计算,并计算完momap输入文件中需要的参数后,接下来将使用MOMAP软件对azulene的S1→S0的辐射速率和内转换速率进行计算,通过对比辐射速率和内转换速率来解释azulene的S1→S0的荧光难以被观测到的原因。
首先计算S1→S0的荧光辐射速率,第一步为做电子振动耦合(electron-vibration coupling, EVC)计算,该计算基于量化计算输出的分子振动频率、力常数矩阵,同时在内坐标以及直角坐标系下,计算分子跃迁发生初末态间的模式位移、黄昆因子、重整能以及Duschinsky转动矩阵。
将bdf的S0优化频率计算结果改为 zulene-s0.out ,将S1的计算结果改为 azulene-s1.out ,同时放在EVC计算文件夹中。
EVC计算的输入文件 momap.inp 为:
do_evc = 1
&evc
ffreq(1) = "azulene-s0.out"
ffreq(2) = "azulene-s1.out"
proj_reorg = .t.
/
提交脚本文件 momap.slurm ,任务运行结束后,生成如下文件

其中 evc.cart.dat 为利用直角坐标系计算得到的模式位移、黄昆因子、重整能以及 Duschinsky 转动矩阵的结果;而 evc.dint.dat 为内坐标计算模式位移、黄昆因子、重整能,直角坐标计算Duschinsky转动矩阵的结果。
打开 evc.cart.dat 文件,查看总重整能的数值:
--------------------------------------------------------------------------------------------------------------------------------------------
Total reorganization energy (cm-1): 4032.869339 5126.265767
--------------------------------------------------------------------------------------------------------------------------------------------
并与 evc.dint.dat 文件中的数值进行比较:
--------------------------------------------------------------------------------------------------------------------------------------------
Total reorganization energy (cm-1): 4070.407661 5114.173064
--------------------------------------------------------------------------------------------------------------------------------------------
比较 evc.cart.dat 以及 evc.dint.dat 文件内的重整能,若重整能相差不大(< \(1000 cm^{-1}\) ),使用 evc.cart.dat 文件进行后续计算,若重整能相差较大(一般情况 evc.cart.dat 大于 evc.dint.dat ),使用 evc.dint.dat 文件进行后续计算。这里两者较为接近,且数值较为合理(< \(10000 cm^{-1}\) ),可使用 evc.cart.dat 进行下一步S1→S0的荧光辐射速率计算。
此外,还可以根据EVC计算的结果文件做更多的后处理。
evc.dx.x.com 和 evc.dx.x.xyz 为两个电子态分子叠加图,其中 evc.dx.x.com 可用GaussView打开,在View-Display Format-Molecule中选择Tube类型,显示如下:

evc.dx.x.xyz 可用Jmol软件打开,显示如下:

evc.dx.v.xyz 也是分子态叠加图,用Jmol软件打开,选择显示-矢量-0.1A,显示如下:

evc.cart.abs 为Duschinsky矩阵文件,可用来画Duschinsky矩阵二维图。可以在Device Studio中,选择Simulator-momap-analysis,打开 evc.cart.abs 文件,显示如下:

同样的方法打开 evc.dint.abs 文件,可在ColorType下拉选框选择显示颜色,显示如下:
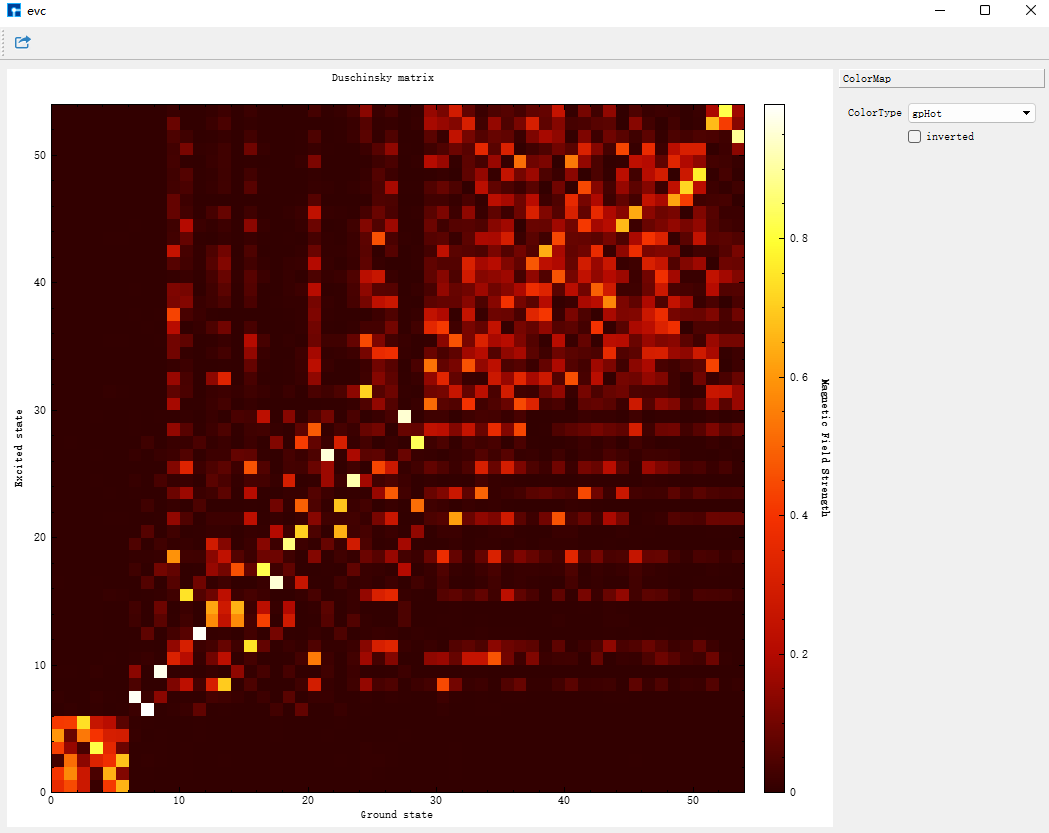
Duschinsky矩阵都是用直角坐标计算的,二者选一即可。
将 evc.vib1.xyz 和 evc.vib2.xyz 文件与 evc.cart.dat 文件放在同一路径下,在Device Studio中,选择Simulator-momap-analysis,打开 evc.cart.dat 文件,出现基态和激发态下各个振动模式对应的重整能和黄昆因子图,显示如下:
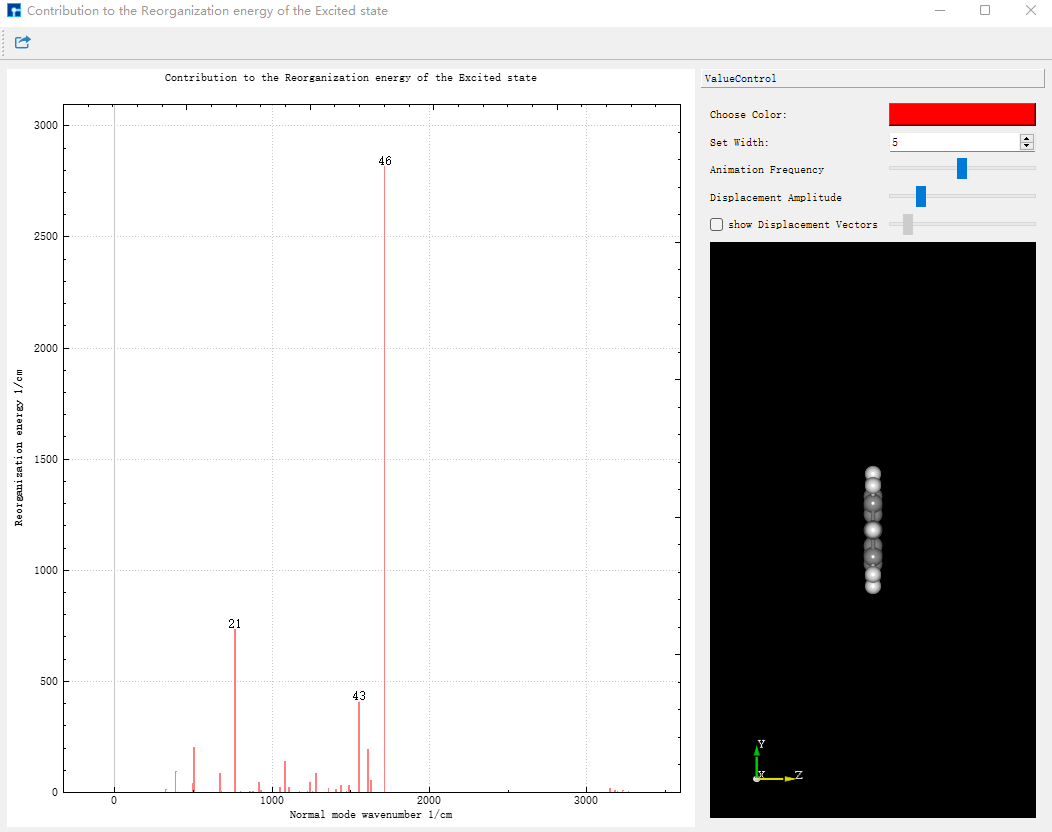
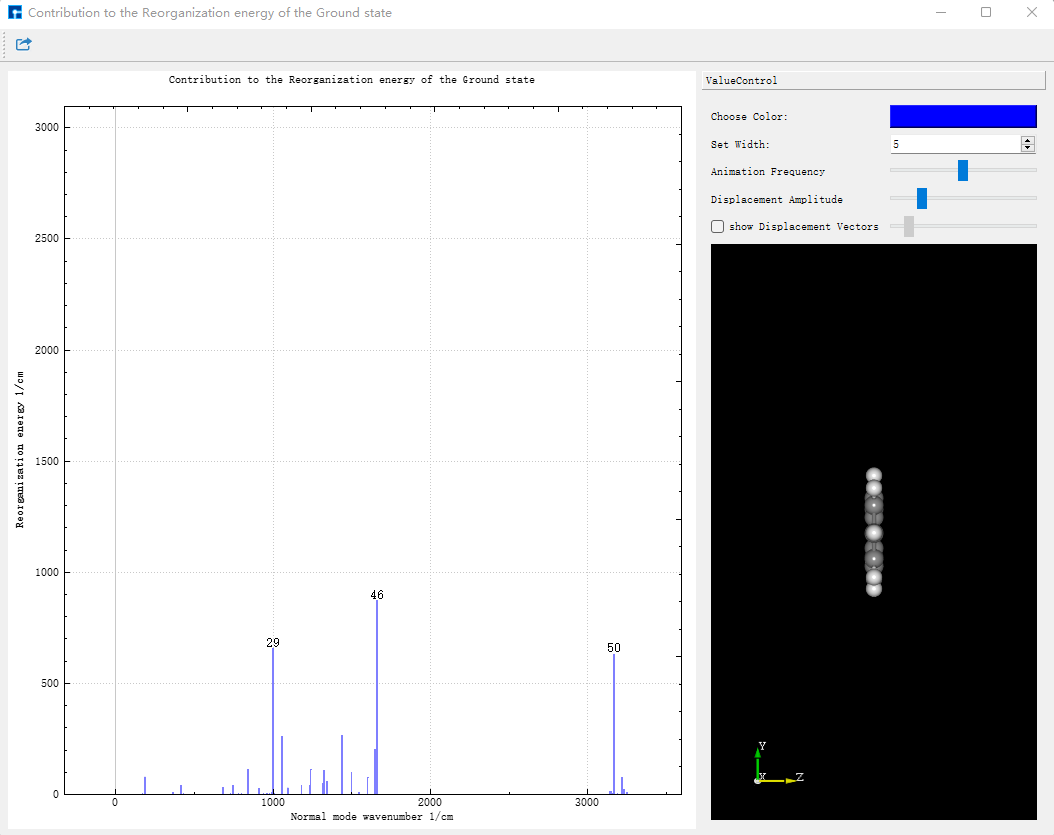

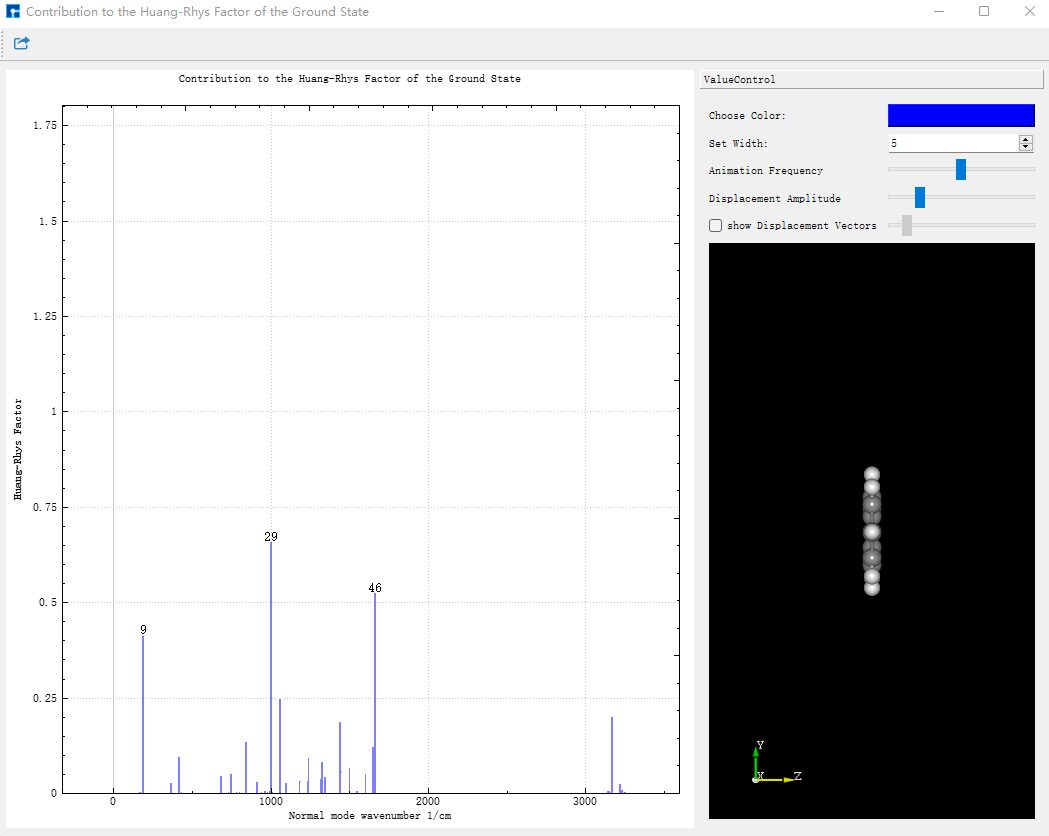
点击Choose Color可以改变线的颜色,改变Set Width中的数值可改变线的粗细。点击图中的线,右侧结构将展示相应的振动模式。振动的快慢可通过Animation Frequency调整,振动的幅度可根据Displacement Amplitude显示。
接下来进行S1→S0的荧光光谱及荧光辐射速率的计算。此部分计算需要EVC计算所得的 evc.*.dat 文件作为输入。如前所述,将 evc.cart.dat 放在荧光光谱及荧光辐射速率的计算文件中,输入文件 momap.inp 为:
do_spec_tvcf_ft = 1
do_spec_tvcf_spec = 1
&spec_tvcf
DUSHIN = .t.
Temp = 300 K
tmax = 1000 fs
dt = 1 fs
Ead = 0.07502 au
EDMA = 0.9267 debye
EDME = 0.6507 debye
FreqScale = 0.70
DSFile = "evc.cart.dat"
Emax = 0.3 au
dE = 0.00001 au
logFile = "spec.tvcf.log"
FtFile = "spec.tvcf.ft.dat"
FoFile = "spec.tvcf.fo.dat"
FoSFile = "spec.tvcf.spec.dat"
/
提交脚本文件 momap.slurm ,任务计算结束后,生成文件如下所示:

spec.tvcf.ft.dat为关联函数文件,内容如下:
#F(t) file
#time(fs) abs_FC_Re abs_FC_Im emi_FC_Re emi_FC_Im
-1000.00000 -3.222659695E-06 -3.680115014E-05 -3.738401821E-06 1.830624064E-05
-999.00000 -1.314545717E-05 -4.054982998E-05 -1.645610065E-07 1.674654956E-05
-998.00000 -2.552186539E-05 -3.557857053E-05 1.690265080E-06 1.428576477E-05
-997.00000 -3.030022340E-05 -2.305671104E-05 2.187359504E-06 1.222900063E-05
-996.00000 -2.620263828E-05 -1.307399386E-05 2.137378198E-06 1.094934359E-05
-995.00000 -1.984646799E-05 -8.898814274E-06 2.126481370E-06 1.032271509E-05
-994.00000 -1.480939587E-05 -8.192889658E-06 2.544875562E-06 9.992907379E-06
-993.00000 -1.150600369E-05 -8.808752896E-06 3.437467322E-06 9.316576838E-06
-992.00000 -9.618163543E-06 -9.696518247E-06 4.080694570E-06 7.874673573E-06
-991.00000 -8.767387663E-06 -1.012196928E-05 3.827322982E-06 6.315202077E-06
-990.00000 -8.083181010E-06 -9.635894264E-06 3.049519176E-06 5.302908607E-06
-989.00000 -6.767527992E-06 -8.787573767E-06 2.230010837E-06 4.817300527E-06
-988.00000 -4.968246898E-06 -8.254624823E-06 1.540853782E-06 4.667945882E-06
-987.00000 -3.046038369E-06 -8.166135596E-06 1.028297048E-06 4.730444119E-06
-986.00000 -1.166755532E-06 -8.469415398E-06 7.338287121E-07 4.901384390E-06
计算完成后先确认关联函数是否收敛,将 spec.tvcf.ft.dat 拖入origin中,选择第一列和第二列作图:

随时间趋于0表示吸收光谱计算收敛。选择第一列和第四列作图:
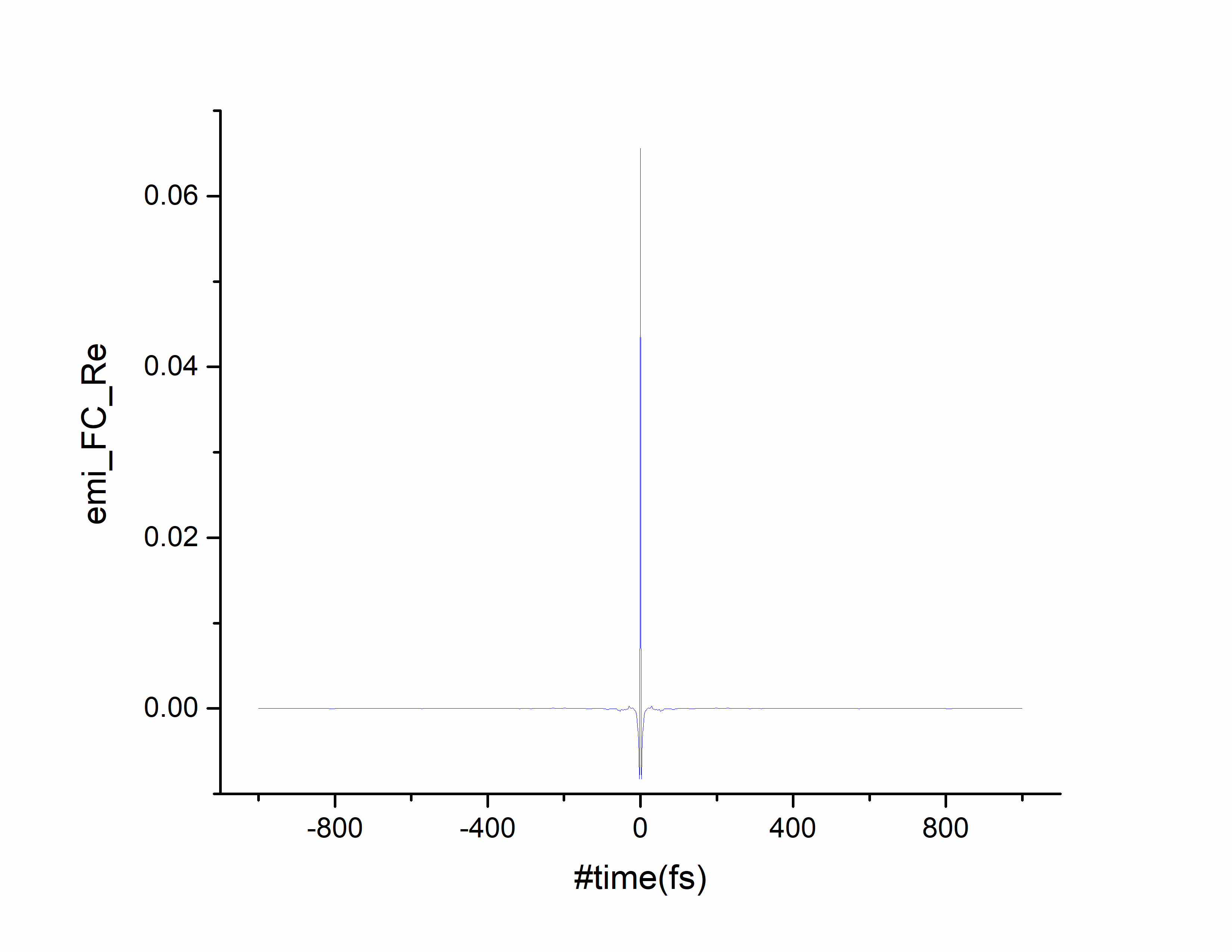
随时间趋于0表示发射光谱计算收敛。
spec.tvcf.spec.dat 为光谱文件:
#spectrum
#1Energy(Hartree) 2Energy(eV) 3WaveNumber(cm-1) 4WaveLength(nm) 5FC_abs 6FC_emi 7FC_emi_intensity
1.63970997E-05 4.46187977E-04 3.59874741E+00 2.77874462E+06 2.22315237E-05 1.35978424E-12 5.71912365E-20
9.23885920E-05 2.51402258E-03 2.02769521E+01 4.93170766E+05 1.24781305E-04 2.44296355E-10 5.78932299E-17
1.68380084E-04 4.58185718E-03 3.69551568E+01 2.70598229E+05 2.26516688E-04 1.48614131E-09 6.41864375E-16
2.44371577E-04 6.64969178E-03 5.36333615E+01 1.86451114E+05 3.27482697E-04 4.56299919E-09 2.86018106E-15
3.20363069E-04 8.71752639E-03 7.03115662E+01 1.42224111E+05 4.27602118E-04 1.03315707E-08 8.48987397E-15
3.96354561E-04 1.07853610E-02 8.69897710E+01 1.14956045E+05 5.27021609E-04 1.96507539E-08 1.99781616E-14
4.72346054E-04 1.28531956E-02 1.03667976E+02 9.64618045E+04 6.25550501E-04 3.34234748E-08 4.04952733E-14
5.48337546E-04 1.49210302E-02 1.20346180E+02 8.30936218E+04 7.23426877E-04 5.25150757E-08 7.38625716E-14
6.24329038E-04 1.69888648E-02 1.37024385E+02 7.29797108E+04 8.20443555E-04 7.78979043E-08 1.24747472E-13
7.00320530E-04 1.90566994E-02 1.53702590E+02 6.50607125E+04 9.16771962E-04 1.10429551E-07 1.98369368E-13
7.76312023E-04 2.11245340E-02 1.70380794E+02 5.86920611E+04 1.01226209E-03 1.51170543E-07 3.01020401E-13
8.52303515E-04 2.31923686E-02 1.87058999E+02 5.34590693E+04 1.10713559E-03 2.00942496E-07 4.39297283E-13
9.28295007E-04 2.52602032E-02 2.03737204E+02 4.90828371E+04 1.20107277E-03 2.60943515E-07 6.21333781E-13
1.00428650E-03 2.73280378E-02 2.20415409E+02 4.53688790E+04 1.29454751E-03 3.31900017E-07 8.54982733E-13
1.08027799E-03 2.93958724E-02 2.37093613E+02 4.21774330E+04 1.38703730E-03 4.15196637E-07 1.15048722E-12
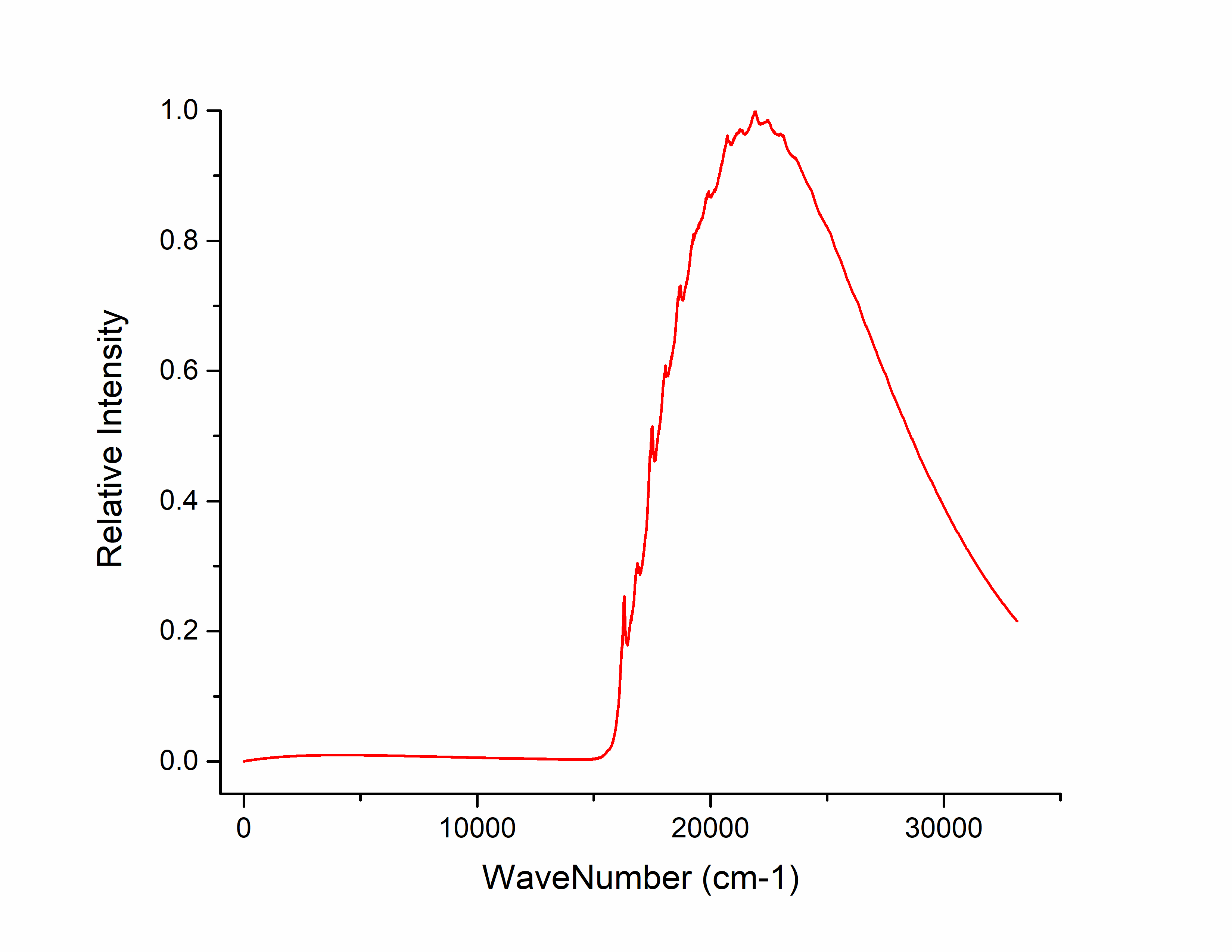
将 spec.tvcf.spec.dat 拖入origin中,选择第三列和第五列作图,得到吸收光谱:
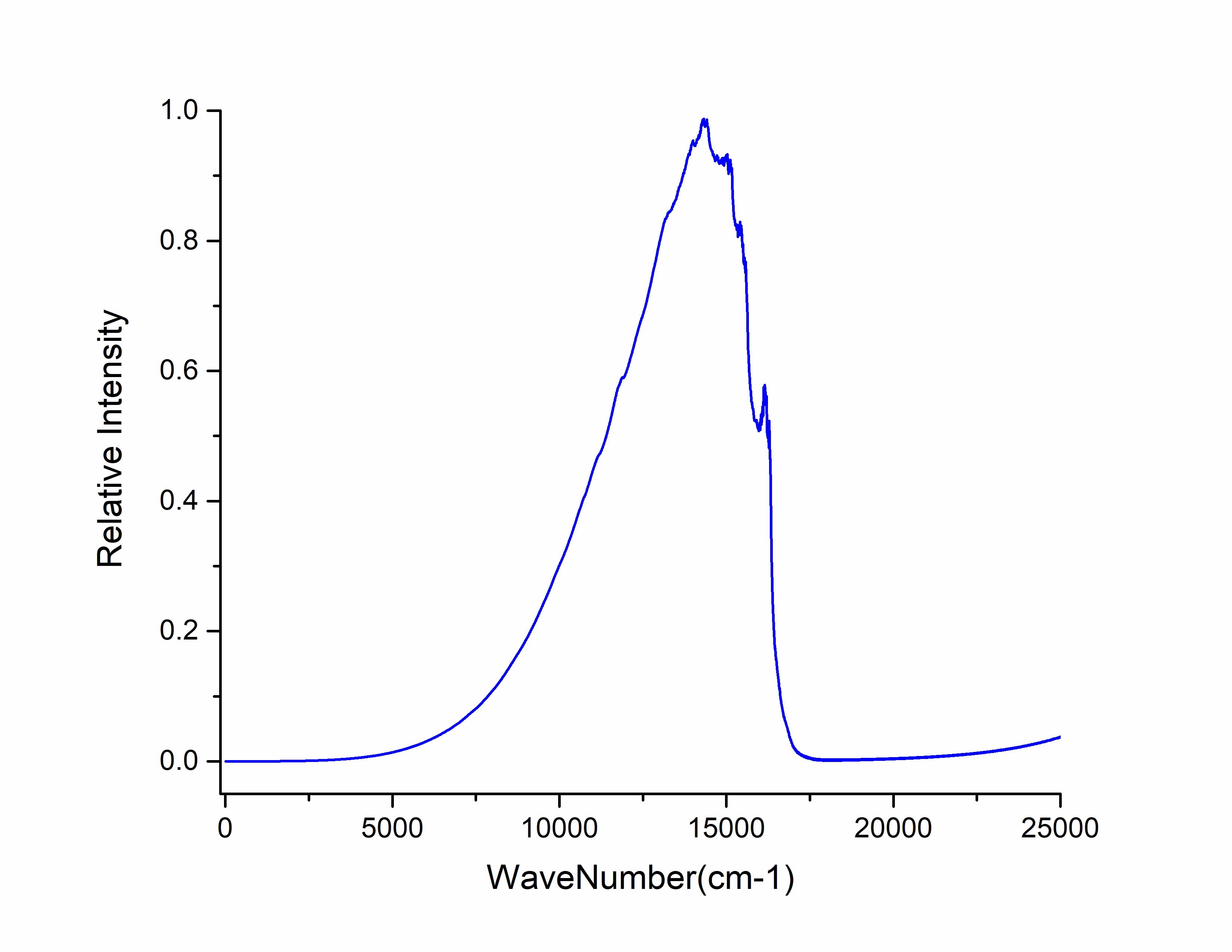
选择第三列和第六列作图,得到发射光谱:
打开spec.tvcf.log,文件末尾输出了荧光辐射速率值,
radiative rate (0): 7.21227543E-12 2.98165371E+05 /s, 3353.84 ns
荧光辐射速率在第一个数和第二个数读取,单位分别为a.u.和 \(s^{-1}\) ,第三个数为寿命,单位为ns。这里,azulene的S1→S0的荧光辐射速率为 2.98165371E+05 /s,荧光寿命为3353.84 ns。
接下来计算azulene的内转换速率。第一步为EVC振动分析计算。
内转换速率的EVC振动分析计算需要非绝热耦合计算结果文件。将非绝热耦合计算结果文件改名为 azulene-nacme.out ,与 azulene-s0.out 和 azulene-s1.out 放在内转换速率计算文件夹下,输入文件 momap.inp 为:
do_evc = 1
&evc
ffreq(1) = "azulene-s0.log"
ffreq(2) = "azulene-s1.log"
fnacme = "azulene-nacme.log"
proj_reorg = .t.
/
提交脚本文件 momap.slurm ,任务计算结束后,生成如下文件:

与前述荧光辐射速率的evc计算相比,多出一个 evc.cart.nac 文件,该文件将和 evc.cart.dat 文件一起用于接下来的内转换速率的计算。
内转换速率输入文件 momap.inp 为:
do_ic_tvcf_ft = 1
do_ic_tvcf_spec = 1
&ic_tvcf
DUSHIN = .t.
Temp = 300 K
tmax = 1000 fs
dt = 1 fs
Ead = 0.07502 au
FreqScale = 0.40
DSFile = "evc.cart.dat"
CoulFile = "evc.cart.nac"
Emax = 0.3 au
logFile = "ic.tvcf.log"
FtFile = "ic.tvcf.ft.dat"
FoFile = "ic.tvcf.fo.dat"
/
计算结束后,生成文件如下:

其中 ic.tvcf.ft.dat 为关联函数文件,内容如下:
#time(fs) IC_ft_Re(au) IC_ft_Im(au)
-1000.00000 -0.302000787E-13 0.332374045E-13
-999.00000 -0.205081112E-13 0.364896559E-13
-998.00000 -0.114899559E-13 0.364092975E-13
-997.00000 -0.403418594E-14 0.342290784E-13
-996.00000 0.187447714E-14 0.309462504E-13
-995.00000 0.652215564E-14 0.270373111E-13
-994.00000 0.100875106E-13 0.226564272E-13
-993.00000 0.125729252E-13 0.178887681E-13
-992.00000 0.138917246E-13 0.128953745E-13
-991.00000 0.139788795E-13 0.793558174E-14
-990.00000 0.128516224E-13 0.332590373E-14
-989.00000 0.106275531E-13 -0.595694484E-15
-988.00000 0.755502051E-14 -0.349394006E-14
-987.00000 0.406056718E-14 -0.511604680E-14
-986.00000 0.703538619E-15 -0.544879345E-14
-985.00000 -0.203569213E-14 -0.479493437E-14
计算结束后首先确认关联函数是否收敛,将 ic.tvcf.ft.dat 拖入origin中,选择第一列和第二列作图:
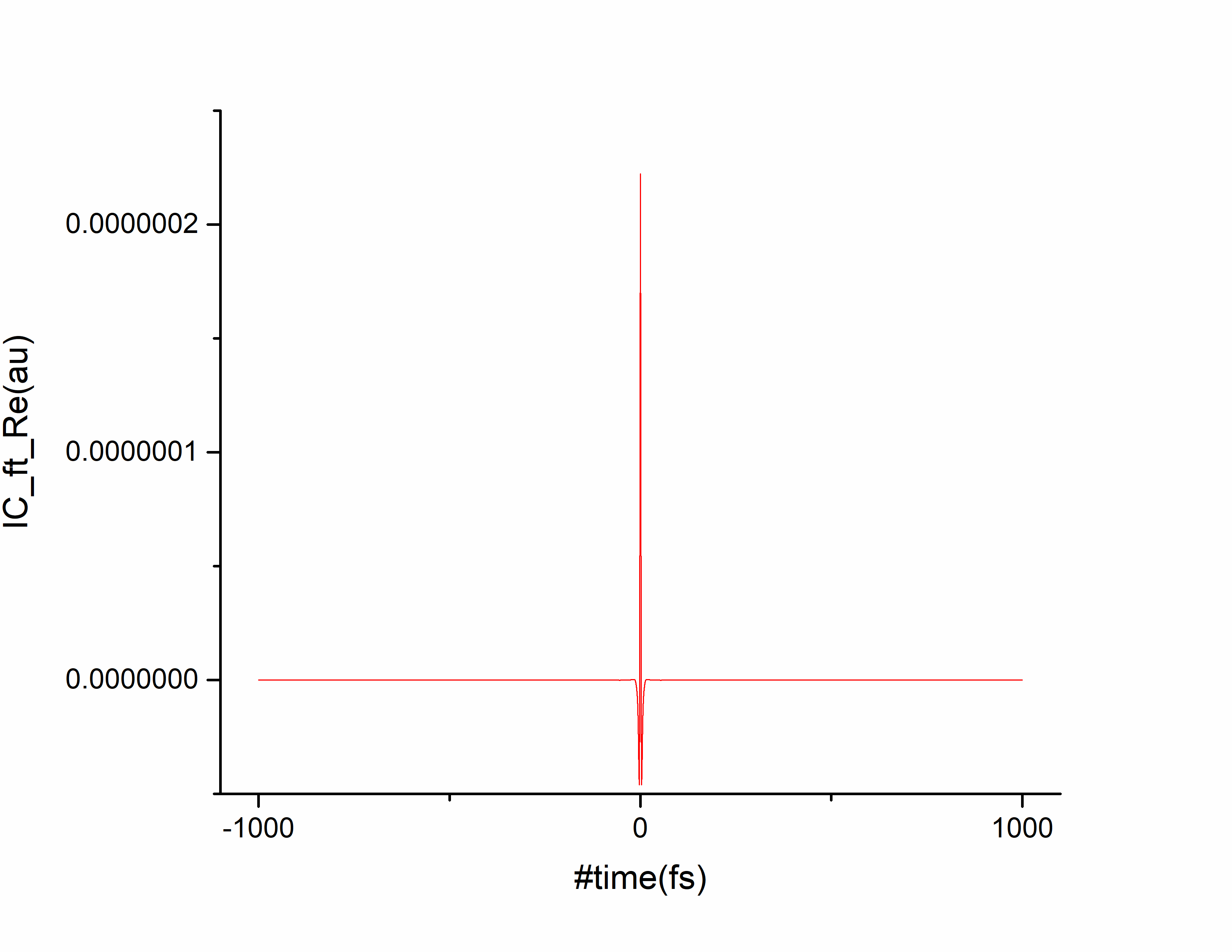
随时间趋于0表示关联函数收敛。
其中 ic.tvcf.fo.dat 为谱函数文件,内容如下:
#1Energy(Hartree) 2Energy(eV) 3WaveNumber(cm-1) 4WaveLength(nm) 5radi-spectrum 6kic(s^{-1}) 7log(kic)
0.00000000E+00 0.00000000E+00 0.00000000E+00 Infinity 0.42121076E-05 0.17413431E+12 0.11240884E+02
0.75991492E-04 0.20678346E-02 0.16678205E+02 0.59958492E+06 0.44127214E-05 0.18242796E+12 0.11261091E+02
0.15198298E-03 0.41356692E-02 0.33356409E+02 0.29979246E+06 0.46209108E-05 0.19103480E+12 0.11281112E+02
0.22797448E-03 0.62035038E-02 0.50034614E+02 0.19986164E+06 0.48375050E-05 0.19998910E+12 0.11301006E+02
0.30396597E-03 0.82713384E-02 0.66712819E+02 0.14989623E+06 0.50633471E-05 0.20932572E+12 0.11320823E+02
0.37995746E-03 0.10339173E-01 0.83391024E+02 0.11991698E+06 0.52978153E-05 0.21901896E+12 0.11340482E+02
0.45594895E-03 0.12407008E-01 0.10006923E+03 0.99930820E+05 0.55357841E-05 0.22885692E+12 0.11359564E+02
0.53194045E-03 0.14474842E-01 0.11674743E+03 0.85654988E+05 0.57737058E-05 0.23869293E+12 0.11377840E+02
0.60793194E-03 0.16542677E-01 0.13342564E+03 0.74948115E+05 0.60096238E-05 0.24844610E+12 0.11395232E+02
为检查是否满足能隙定律,将 ic.tvcf.fo.dat 拖入origin中,选择第三列和第七列作图:
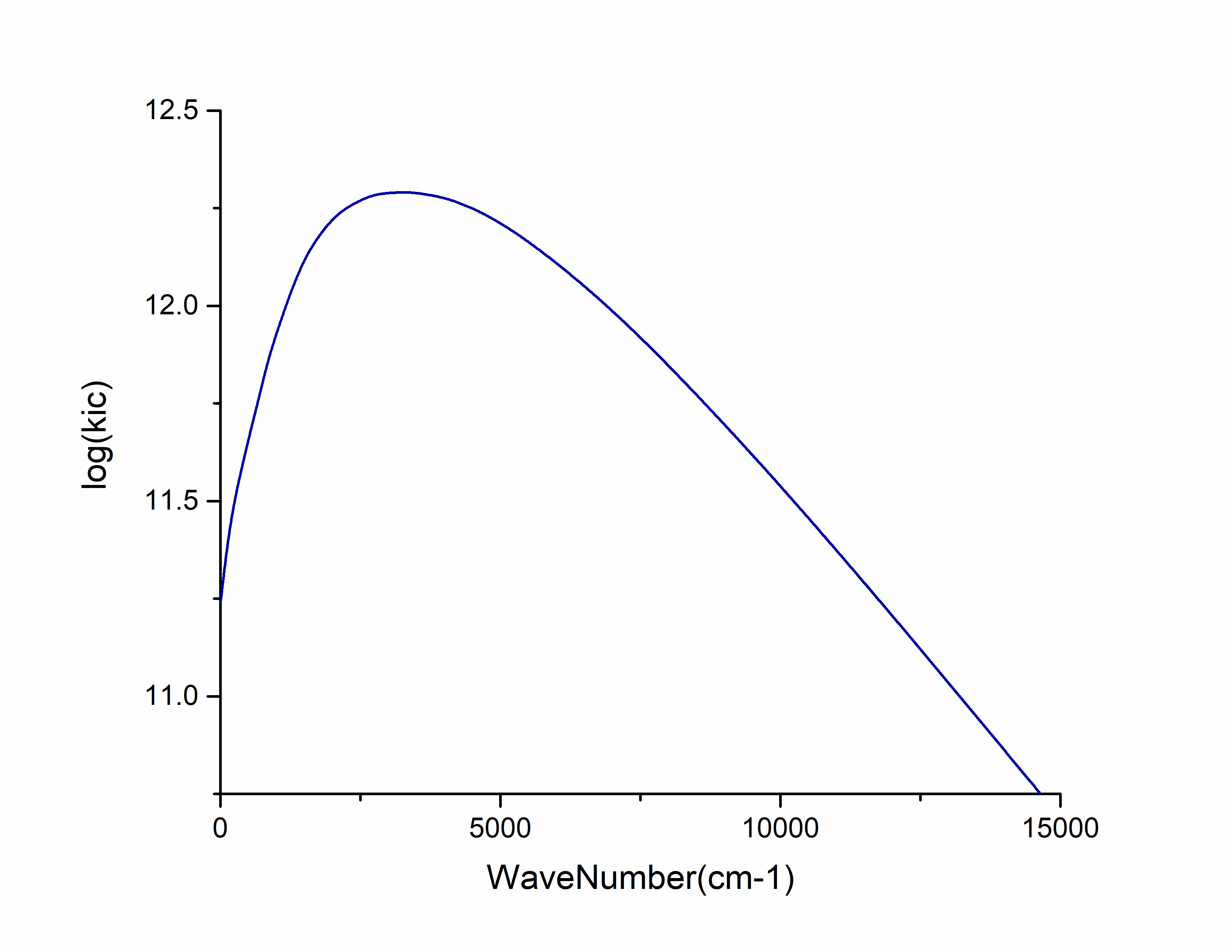
此外, ic.tvcf.fo.dat 文件中第一列和第六列表示不同Ead下的非辐射速率。
在 ic.tvcf.log 文件的末尾,读取S1→S0的非辐射速率值,
#1Energy(Hartree) 2Energy(eV) 3WaveNumber(cm-1) 4WaveLength(nm) 5radi-spectrum 6kic(s^{-1}) 7log(kic) 8time(ps)
7.50036029E-02 2.04095275E+00 1.64613880E+04 6.07482186E+02 6.54396018E-07 2.70536301E+10 1.04322255E+01 36.96361624
这里S1→S0的非辐射速率为2.70536301E+10 \(s^{-1}\) 。
对比azulene的荧光辐射速率和非辐射速率,荧光辐射速率为2.98165371E+05 /s,非辐射速率为2.70536301E+10 \(s^{-1}\) ,非辐射速率比荧光辐射速率高五个数量级,从而降低了azulene的S1→S0的荧光量子效率,所以S1→S0的荧光难以被观测到,从而表现出反kasha规则的荧光现象。