含时密度泛函理论
BDF支持多种激发态计算方法,可用于UV-Vis/XAS吸收光谱、荧光光谱/速率/寿命/量子产率、磷光光谱/速率/寿命/量子产率、内转换(IC)速率、系间窜越(ISC)速率等的计算,其中以基于Kohn-Sham参考态的线性响应含时密度泛函 (TDDFT)方法,以及TDDFT方法的Tamm-Dancoff近似(TDA)为主。其中涉及速率、寿命、量子产率及振动分辨光谱的计算,BDF仅负责计算所需的平衡结构、Hessian、跃迁矩阵元等计算,后续计算需要结合MOMAP程序完成;但垂直吸收/垂直发射光谱(即不关心光谱振动精细结构的情形)可以仅用BDF程序计算得到。与其他量化软件相比,BDF的TDDFT模块独具特色,主要体现在:
支持各种自旋翻转(spin-flip)方法;
支持自旋匹配TDDFT方法X-TDDFT,可以有效解决参考态为开壳层时激发态存在自旋污染的问题,适用于自由基、过渡金属等体系的激发态计算;
支持芯激发态(core excited state)相关的计算,如计算X射线吸收谱(XAS)。一般的TDDFT算法为了计算一个激发态,常需同时把比该激发态激发能更低的所有态均计算出来,而芯激发态的能量通常很高,这样计算效率太低。BDF所用的iVI方法则可在不计算更低的激发态的情况下,直接计算某个较高的能量区间内的所有激发态,从而节省计算资源;
支持一阶非绝热耦合矩阵元(first-order non-adiabatic coupling matrix element, fo-NACME,或简称NACME)的计算,尤其是激发态和激发态之间的NACME。NACME主要用于研究非辐射跃迁过程,如结合MOMAP软件用费米黄金规则计算内转换速率常数(见 示例 ),或用非绝热动力学研究内转换、光化学反应的过程等等。很多量子化学程序支持基态和激发态之间的NACME,但支持激发态和激发态之间的NACME的程序较少,因此对于激发态到激发态的内转换以及多态光化学反应等过程,BDF相比现有大部分量子化学程序有独特的优势。
除TDDFT之外,BDF还支持利用 mom方法 在SCF水平下计算激发态。
危险
所有 SCAN 家族的泛函(如SCAN0,r2SCAN)都存在“三重态不稳定”问题 [69] , 不要用于TDDFT自旋翻转计算(例如对闭壳层体系计算三重激发态)。这种情况推荐用TDA。
闭壳层体系计算:R-TDDFT
R-TDDFT用于计算闭壳层体系。如果基态计算从RHF出发,TDDFT模块执行的是TDHF计算。 利用TDDFT计算 \(\ce{H2O}\) 分子激发能,简洁输入如下:
#!bdf.sh
TDDFT/B3lyp/cc-pvdz iroot=1
geometry
O
H 1 R1
H 1 R1 2 109.
R1=1.0 # OH bond length in angstrom
end geometry
这里,关键词 TDDFT/B3lyp/cc-pvdz 指定执行TDDFT计算,所用泛函为 B3lyp ,基组为 cc-pVDZ 。
与之对应的高级输入为:
$compass
Geometry
O
H 1 1.0
H 1 1.0 2 109.
End geometry
Basis
cc-pvdz
$end
$xuanyuan
$end
$scf
RKS # Restricted Kohn-sham
DFT # DFT exchange-correlation functional B3lyp
b3lyp
$end
# input for tddft
$tddft
iroot # For each irrep, calculate 1 root.
1 #on default, 10 roots are calculated for each irreps if advanced input used
$end
完成计算将顺序调用 COMPASS , XUANYUAN , SCF 及 TDDFT 四个模块。其中 SCF 模块执行RKS计算。 基于RKS的计算结果,进行后续的 TDDFT 计算。
注意因为水分子属于 \(\rm C_{2v}\) 点群,共有4个不可约表示,而不同不可约表示的激发态是分别求解的,因此视用户需求而定,有以下若干种指定激发态数目的方法,例如:
(1)每个不可约表示均计算1个激发态:
$TDDFT
iroot
1
$END
此时每个不可约表示计算得到的激发态大概率是该不可约表示下能量最低的激发态,但是这一点无法保证,也就是说有较小的概率会收敛到第二激发态甚至更高的某个激发态。如果要提高得到最低激发态的概率,可以写
$TDDFT
iroot
2
$END
此时每个不可约表示计算2个激发态,且每个不可约表示下计算得到的第一个激发态是该不可约表示下能量最低的激发态的概率较iroot=1时更高。此外,此时每个不可约表示下计算得到的第二个激发态大概率是该不可约表示下能量第二低的激发态,但满足这一点的概率较“计算得到的第一个激发态是该不可约表示下能量最低的激发态”的概率更低。如果进一步增加iroot,则计算得到的第一个激发态是能量最低的激发态的概率很快趋近于100%,但永远无法严格达到100%。
出于类似的原因,不仅当计算1个激发态时常常需要将iroot设为大于1,当计算N(N>1)个激发态时,若想相对可靠地确保这N个激发态是能量最低的N个激发态,也需要将iroot设为大于N。一般而言,当分子满足下述条件之一时,应当将iroot设得较大,例如比所需的激发态数目大至少3个:(1)分子具有近似的点群对称性;(2)分子虽然具有精确的点群对称性,但是受程序限制或者应用户需要,计算在更低的点群下进行,例如在开壳层TDDFT(见下文)计算中,因开壳层TDDFT代码不支持非阿贝尔点群,而改为在最大的阿贝尔子群下进行计算。当分子不属于上述情况之一时,iroot只需比所需的激发态数目略大即可,例如大1~2个。
(2)只计算一个B1激发态和一个B2激发态,不计算其他不可约表示下的激发态:
#! tdtest.sh
TDDFT/B3lyp/3-21G nroot=0,0,1,1
Geometry
...
End geometry
或者
$TDDFT
nroot
0 0 1 1 # 也可输入为 0,0,1,1
$END
其中nroot关键词表明用户分别对每个不可约表示指定激发态的数目。因程序内部将 \(\rm C_{2v}\) 点群的不可约表示以A1、A2、B1、B2的顺序排列(见点群相关章节关于各个不可约表示的排序的介绍),因此以上输入表明只计算B1、B2各一个激发态。类似iroot的情形,如需要相对可靠地确保计算得到的是相应不可约表示下能量最低的态,则应当将nroot设得比所需值略大。
(3)计算最低的4个激发态,而不限定这些激发态的不可约表示
#! tdtest.sh
TDDFT/B3lyp/3-21G iroot=-4
Geometry
...
End geometry
或者
$TDDFT
iroot
-4
$END
此时程序通过初始猜测的激发能来判断各个不可约表示应当求解多少个激发态,但因为初始猜测的激发能排列顺序可能和完全收敛的激发能有一定差异,程序不能严格保证求得的4个激发态一定是能量最低的4个激发态。如用户要求严格保证得到的4个激发态为最低的4个激发态,用户应当令程序计算多于4个激发态,如8个激发态,然后取能量最低的4个。
Kohn-Sham计算的输出前面已经介绍过,这里我们只关注 TDDFT 计算的结果。程序输出会先给出TDDFT计算的设置信息,方便用户检查是否计算的设置,如下:
--------------------------------------------------
--- PRINT: Information about TDDFT calculation ---
--------------------------------------------------
ERI Maxblk= 8M
[print level]
iprt= 0
[method]
R-TD-DFT
isf= 0
SC Excitations
RPA: (A-B)(A+B)Z=w2*Z
[special choice for method]
ialda= 0
[active space]
Full active space
[algorithm]
Target Excited State in each rep / Diag method :
1 A1 1 1
2 A2 1 1
3 B1 1 1
4 B2 1 1
[dvdson_parameters]
iupdate = 3
Nfac = 50
Nmaxcycle= 50
nblock = 50
crit_e = 0.10E-06
crit_vec = 0.10E-04
crit_demo= 0.10E-07
crit_indp= 0.10E-09
guess = 20
dump = 0
[output eigenvector control]
cthrd= 0.100
-------------------------------------------------
--- END : Information about TDDFT calculation ---
-------------------------------------------------
这里,
R-TD-DFT表示正在进行的是基于限制性基态波函数计算的TDDFT;isf= 0表示计算不翻转自旋;ialda= 0表示使用Full non-collinear Kernel,这是非自旋翻转TDDFT的默认Kernel。
下面的输出给出了每个不可约表示计算的根的数目。
Target Excited State in each rep / Diag method :
1 A1 1 1
2 A2 1 1
3 B1 1 1
4 B2 1 1
TDDFT模块还会打印占据轨道,虚轨道等TDDFT计算的活性轨道信息
Print [Active] Orbital List
---[Alpha set]---
idx irep (rep,ibas,type) F_av(eV) iact
---------------------------------------------------
1 1 A1 1 2 -520.34813 0.05
2 1 A1 2 2 -26.42196 1.84
3 3 B1 1 2 -13.66589 2.96
4 1 A1 3 2 -9.50404 2.49
5 4 B2 1 2 -7.62124 2.12
6 1 A1 4 0 1.23186 9.86
7 3 B1 2 0 3.27539 11.48
8 3 B1 3 0 15.02893 7.40
9 1 A1 5 0 15.44682 6.60
10 1 A1 6 0 24.53525 4.35
11 4 B2 2 0 25.07569 3.88
12 3 B1 4 0 27.07545 6.17
13 2 A2 1 0 33.09515 3.99
14 1 A1 7 0 34.03695 5.08
15 4 B2 3 0 39.36812 4.67
16 3 B1 5 0 43.83066 4.86
17 1 A1 8 0 43.91179 4.34
18 3 B1 6 0 55.56126 4.35
19 1 A1 9 0 56.13188 4.04
20 4 B2 4 0 78.06511 2.06
21 2 A2 2 0 80.16952 2.10
22 1 A1 10 0 83.17934 2.38
23 1 A1 11 0 94.37171 2.81
24 3 B1 7 0 99.90789 2.86
这里,轨道1-5是占据轨道,6-24是虚轨道,其中,第5个和第6个轨道分别是HOMO和LUMO轨道, 分别属于不可约表示B2和不可约表示A1, 轨道能分别是-7.62124 eV和1.23186 eV。由于 \(\ce{H2O}\) 分子有4个不可约表示,TDDFT会对每个不可约表示逐一求解。 在进入Davidson迭代求解Casida方程之前,系统会估计内存使用情况,
==============================================
Jrep: 1 ExctSym: A1 (convert to td-psym)
Irep: 1 PairSym: A1 GsSym: A1
Nexit: 1 Nsos: 33
==============================================
Estimated memory for JK operator: 0.053 M
Maxium memory to calculate JK operator: 512.000 M
Allow to calculate 1 roots at one pass for RPA ...
Allow to calculate 2 roots at one pass for TDA ...
Nlarge= 33 Nlimdim= 33 Nfac= 50
Estimated mem for dvdson storage (RPA) = 0.042 M 0.000 G
Estimated mem for dvdson storage (TDA) = 0.017 M 0.000 G
这里,系统统计存储JK算符需要的内存约 0.053MB, 输入设置的内存是512MB (见 memjkop 关键词 )。
系统提示RPA计算,即完全的TDDFT计算每次(one pass)可以算1个根,TDA计算每次可以算2个根。由于分子体系小,内存足够。
分子体系较大时,如果这里输出的允许的每次可算根的数目小于系统设置数目,TDDFT模块将根据最大允许可算根的数目,通过
多次积分计算构造JK算符,导致计算效率降低,用户需要用 memjkop 关键词增加内存。
Davidson迭代开始计算输出信息如下,
Iteration started !
Niter= 1 Nlarge = 33 Nmv = 2
Ndim = 2 Nlimdim= 33 Nres= 31
Approximated Eigenvalue (i,w,diff/eV,diff/a.u.):
1 9.5246226546 9.5246226546 0.350E+00
No. of converged eigval: 0
Norm of Residuals:
1 0.0120867135 0.0549049429 0.121E-01 0.549E-01
No. of converged eigvec: 0
Max norm of residues : 0.549E-01
*** New Directions : sTDDFT-Davidson step ***
Left Nindp= 1
Right Nindp= 1
Total Nindp= 2
[tddft_dvdson_ZYNI]
Timing For TDDFT_AVmat, Total: 0.08s 0.02s 0.02s
MTrans1: 0.00s 0.02s 0.00s
COULPOT: 0.00s 0.00s 0.00s
AVint : 0.08s 0.00s 0.02s
MTrans2: 0.00s 0.00s 0.00s
TDDFT ZYNI-AV time-TOTAL 0.08 S 0.02 S 0.02 S
TDDFT ZYNI-AV time-Coulp 0.08 S 0.02 S 0.02 S
TDDFT ZYNI-AV time-JKcon 0.00 S 0.00 S 0.00 S
tddft JK operator time: 0.00 S 0.00 S 0.00 S
Niter= 2 Nlarge = 33 Nmv = 4
Ndim = 4 Nlimdim= 33 Nres= 29
Approximated Eigenvalue (i,w,diff/eV,diff/a.u.):
1 9.3817966321 0.1428260225 0.525E-02
No. of converged eigval: 0
Norm of Residuals:
1 0.0029082582 0.0074085379 0.291E-02 0.741E-02
No. of converged eigvec: 0
收敛信息如下:
Niter= 5 Nlarge = 33 Nmv = 10
Ndim = 10 Nlimdim= 33 Nres= 23
Approximated Eigenvalue (i,w,diff/eV,diff/a.u.):
1 9.3784431931 0.0000001957 0.719E-08
No. of converged eigval: 1
### Cong: Eigenvalues have Converged ! ###
Norm of Residuals:
1 0.0000009432 0.0000023006 0.943E-06 0.230E-05
No. of converged eigvec: 1
Max norm of residues : 0.230E-05
### Cong. Residuals Converged ! ###
------------------------------------------------------------------
Orthogonality check2 for iblock/dim = 0 1
Averaged nHxProd = 10.000
Ndim = 1 Maximum nonzero deviation from Iden = 0.333E-15
------------------------------------------------------------------
------------------------------------------------------------------
Statistics for [dvdson_rpa_block]:
No. of blocks = 1
Size of blocks = 50
No. of eigens = 1
No. of HxProd = 10 Averaged = 10.000
Eigenvalues (a.u.) =
0.3446513056
------------------------------------------------------------------
从上面输出的第一行可以看出,5次迭代计算收敛。系统随后打印收敛后电子态的信息,
No. 1 w=9.3784 eV -76.0358398606 a.u. f= 0.0767 D<Pab>= 0.0000 Ova= 0.5201
CV(0): A1( 3 )-> A1( 4 ) c_i: 0.9883 Per: 97.7% IPA: 10.736 eV Oai: 0.5163
CV(0): B1( 1 )-> B1( 2 ) c_i: -0.1265 Per: 1.6% IPA: 16.941 eV Oai: 0.6563
Estimate memory in tddft_init mem: 0.001 M
其中第1行的信息,
No. 1 w= 9.3784 eV表示第一激发态激发能为9.3784 eV;-76.0358398606 a.u.给出第一激发态的总能量;f= 0.0767给出第一激发态与基态之间跃迁的振子强度;D<Pab>= 0.0000为激发态的<S^2>值与基态的<S^2>值之差(对于自旋守恒跃迁,该值反映了激发态的自旋污染程度;对于自旋翻转跃迁,该值与理论值S(S+1)(激发态)-S(S+1)(基态)之差反映了激发态的自旋污染程度);Ova= 0.5201为绝对重叠积分(absolute overlap integral,取值范围为[0,1],该值越接近0,说明相应的激发态的电荷转移特征越明显,否则说明局域激发特征越明显)。
第2行和第3行给出激发主组态信息
CV(0):中CV(0)表示该激发是Core到Virtual轨道激发,0表示是Singlet激发;A1( 3 )-> A1( 4 )给出了电子跃迁的占据-空轨道对,电子从A1表示的第3个轨道跃迁到A1表示的第4个轨道,结合上面输出轨道信息,可看出这是HOMO-2到LUMO的激发;c_i: 0.9883表示该跃迁在整个激发态里的线性组合系数为0.9883;Per: 97.7%表示该激发组态占97.7%;IPA: 10.736 eV代表该跃迁所涉及的两个轨道的能量差为10.736 eV;Oai: 0.5163表示假如该激发态只有这一个跃迁的贡献,那么该激发态的绝对重叠积分为0.5001,由这一信息可以方便地得知哪些跃迁是局域激发,哪些跃迁是电荷转移激发。
所有不可约表示求解完后,所有的激发态会按照能量高低排列总结输出,并打印对应的振子强度等信息,
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 B2 1 B2 7.1935 eV 172.36 nm 0.0188 0.0000 99.8% CV(0): B2( 1 )-> A1( 4 ) 8.853 0.426 0.0000
2 A2 1 A2 9.0191 eV 137.47 nm 0.0000 0.0000 99.8% CV(0): B2( 1 )-> B1( 2 ) 10.897 0.356 1.8256
3 A1 2 A1 9.3784 eV 132.20 nm 0.0767 0.0000 97.7% CV(0): A1( 3 )-> A1( 4 ) 10.736 0.520 2.1850
4 B1 1 B1 11.2755 eV 109.96 nm 0.0631 0.0000 98.0% CV(0): A1( 3 )-> B1( 2 ) 12.779 0.473 4.0820
随后还打印了跃迁偶极矩。
*** Ground to excited state Transition electric dipole moments (Au) ***
State X Y Z Osc.
1 -0.0000 -0.3266 0.0000 0.0188 0.0188
2 0.0000 0.0000 0.0000 0.0000 0.0000
3 0.0000 0.0000 0.5777 0.0767 0.0767
4 0.4778 -0.0000 0.0000 0.0631 0.0631
很多情况下,用户并不知道需要计算多少个激发态,只知道需要计算哪个波长范围内的激发态。为此可以用以下写法
$TDDFT
...
iwindow
300 800 nm
$END
代替上述的iroot等关键词,即代表计算300~800 nm范围内的激发态,或者说计算300~800 nm范围内的UV-Vis吸收光谱(也可以eV、cm-1、au为单位指定激发能范围)。但程序只能保证计算的激发态范围大致是在用户所设定的范围,其既不能保证计算出来的所有激发态都在300~800 nm范围内,也不能保证300~800 nm范围内的所有激发态都被计算出来。若要保证计算出来的激发态既不多也不少,刚好涵盖用户指定的波长范围,可采用 iVI方法 。
开壳层体系计算:U-TDDFT
开壳层体系可以用U-TDDFT计算,但其精度不及后文将要提到的X-TDDFT方法,因此采用U-TDDFT的主要意义在于重复其他程序的U-TDDFT结果,若无需重复其他程序的计算结果,一般建议采用X-TDDFT。例如对于 \(\ce{H2O+}\) 离子,先进行UKS计算,然后利用U-TDDFT计算激发态。典型的输入为,
#!bdf.sh
TDDFT/B3lyp/cc-pvdz iroot=4 group=C(1) charge=1
geometry
O
H 1 R1
H 1 R1 2 109.
R1=1.0 # OH bond length in angstrom
end geometry
这里,关键词
iroot=4指定对每个不可约表示计算4个根;charge=1指定体系的电荷为+1;group=C(1)指定强制使用C1点群计算。
与之对应的高级输入为,
$compass
#Notice: The unit of molecular coordinate is angstrom
geometry
O
H 1 R1
H 1 R1 2 109.
R1=1.0 # OH bond length in angstrom
end geometry
basis
cc-pVDZ
group
C(1) # Force to use C1 symmetry
$end
$xuanyuan
$end
$scf
uks
dft
b3lyp
charge
1
spinmulti
2
$end
$tddft
iroot
4
$end
这个输入要注意的几个细节是:
compass模块中利用关键词group强制计算使用C(1)点群;scf模块设置UKS计算,charge为1,spinmulti(自旋多重度,2S+1)=2;tddft模块的iroot设定每个不可约表示算4个根,由于用了C1对称性,计算给出水的阳离子的前四个激发态。
从以下输出可以看出执行的是U-TDDFT计算:
--------------------------------------------------
--- PRINT: Information about TDDFT calculation ---
--------------------------------------------------
ERI Maxblk= 8M
[print level]
iprt= 0
[method]
U-TD-DFT
isf= 0
SC Excitations
RPA: (A-B)(A+B)Z=w2*Z
计算总结输出的4个激发态为,
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 A 2 A 2.1960 eV 564.60 nm 0.0009 0.0024 99.4% CO(bb): A( 4 )-> A( 5 ) 5.955 0.626 0.0000
2 A 3 A 6.3479 eV 195.31 nm 0.0000 0.0030 99.3% CO(bb): A( 3 )-> A( 5 ) 9.983 0.578 4.1520
3 A 4 A 12.0991 eV 102.47 nm 0.0028 1.9312 65.8% CV(bb): A( 4 )-> A( 6 ) 14.637 0.493 9.9032
4 A 5 A 13.3618 eV 92.79 nm 0.0174 0.0004 97.6% CV(aa): A( 4 )-> A( 6 ) 15.624 0.419 11.1659
其中第3激发态的 D<S^2> 值较大,表明存在自旋污染问题。
开壳层体系:X-TDDFT(也称SA-TDDFT)
X-TDDFT是一种自旋匹配TDDFT方法,用于计算开壳层体系。 开壳层体系U-TDDFT三重态耦合的双占据到虚轨道激发态(在BDF中标记为CV(1))存在自旋污染问题,因而其激发能常被低估。X-TDDFT可以用于解决这一问题。考虑 \(\ce{N2+}\) 分子,X-TDDFT的简洁计算输入为:
#! N2+.sh
X-TDDFT/b3lyp/aug-cc-pvtz group=D(2h) charge=1 spinmulti=2 iroot=5
Geometry
N 0.00 0.00 0.00
N 0.00 0.00 1.1164
End geometry
高级输入:
$compass
#Notice: The unit of molecular coordinate is angstrom
Geometry
N 0.00 0.00 0.00
N 0.00 0.00 1.1164
End geometry
basis
aug-cc-pvtz
group
D(2h) # Force to use D2h symmetry
$end
$xuanyuan
$end
$scf
roks # ask for ROKS calculation
dft
b3lyp
charge
1
spinmulti
2
$end
$tddft
iroot
5
$end
这里, SCF 模块要求用 ROKS 方法计算基态, TDDFT 模块将默认采用 X-TDDFT 计算。
激发态输出为,
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 B2u 1 B2u 0.7902 eV 1569.00 nm 0.0017 0.0195 98.6% CO(0): B2u( 1 )-> Ag( 3 ) 3.812 0.605 0.0000
2 B3u 1 B3u 0.7902 eV 1569.00 nm 0.0017 0.0195 98.6% CO(0): B3u( 1 )-> Ag( 3 ) 3.812 0.605 0.0000
3 B1u 1 B1u 3.2165 eV 385.46 nm 0.0378 0.3137 82.6% CO(0): B1u( 2 )-> Ag( 3 ) 5.487 0.897 2.4263
4 B1u 2 B1u 8.2479 eV 150.32 nm 0.0008 0.9514 48.9% CV(1): B2u( 1 )-> B3g( 1 ) 12.415 0.903 7.4577
5 Au 1 Au 8.9450 eV 138.61 nm 0.0000 1.2618 49.1% CV(0): B2u( 1 )-> B2g( 1 ) 12.903 0.574 8.1548
6 Au 2 Au 9.0519 eV 136.97 nm 0.0000 1.7806 40.1% CV(1): B3u( 1 )-> B3g( 1 ) 12.415 0.573 8.2617
7 B1u 3 B1u 9.0519 eV 136.97 nm 0.0000 1.7806 40.1% CV(1): B3u( 1 )-> B2g( 1 ) 12.415 0.906 8.2617
8 B2g 1 B2g 9.4442 eV 131.28 nm 0.0000 0.0061 99.0% OV(0): Ag( 3 )-> B2g( 1 ) 12.174 0.683 8.6540
9 B3g 1 B3g 9.4442 eV 131.28 nm 0.0000 0.0061 99.0% OV(0): Ag( 3 )-> B3g( 1 ) 12.174 0.683 8.6540
10 Au 3 Au 9.5281 eV 130.12 nm 0.0000 0.1268 37.0% CV(0): B3u( 1 )-> B3g( 1 ) 12.903 0.574 8.7379
11 B1u 4 B1u 9.5281 eV 130.12 nm 0.0000 0.1267 37.0% CV(0): B2u( 1 )-> B3g( 1 ) 12.903 0.909 8.7379
12 Au 4 Au 10.7557 eV 115.27 nm 0.0000 0.7378 49.1% CV(1): B3u( 1 )-> B3g( 1 ) 12.415 0.575 9.9655
13 B3u 2 B3u 12.4087 eV 99.92 nm 0.0983 0.1371 70.4% CV(0): B1u( 2 )-> B2g( 1 ) 15.288 0.793 11.6185
14 B2u 2 B2u 12.4087 eV 99.92 nm 0.0983 0.1371 70.4% CV(0): B1u( 2 )-> B3g( 1 ) 15.288 0.793 11.6185
15 B1u 5 B1u 15.9005 eV 77.98 nm 0.7766 0.7768 32.1% CV(0): B3u( 1 )-> B2g( 1 ) 12.903 0.742 15.1103
16 B2u 3 B2u 17.6494 eV 70.25 nm 0.1101 0.4841 92.0% CV(0): B2u( 1 )-> Ag( 4 ) 19.343 0.343 16.8592
17 B3u 3 B3u 17.6494 eV 70.25 nm 0.1101 0.4841 92.0% CV(0): B3u( 1 )-> Ag( 4 ) 19.343 0.343 16.8592
18 Ag 2 Ag 18.2820 eV 67.82 nm 0.0000 0.0132 85.2% OV(0): Ag( 3 )-> Ag( 4 ) 19.677 0.382 17.4918
19 B2u 4 B2u 18.5465 eV 66.85 nm 0.0021 1.5661 77.8% CV(1): B2u( 1 )-> Ag( 4 ) 19.825 0.401 17.7562
20 B3u 4 B3u 18.5465 eV 66.85 nm 0.0021 1.5661 77.8% CV(1): B3u( 1 )-> Ag( 4 ) 19.825 0.401 17.7562
21 Ag 3 Ag 18.7805 eV 66.02 nm 0.0000 0.2156 40.4% CV(0): B3u( 1 )-> B3u( 2 ) 20.243 0.337 17.9903
22 B1g 1 B1g 18.7892 eV 65.99 nm 0.0000 0.2191 40.5% CV(0): B2u( 1 )-> B3u( 2 ) 20.243 0.213 17.9990
23 B1g 2 B1g 18.8704 eV 65.70 nm 0.0000 0.2625 41.8% CV(0): B3u( 1 )-> B2u( 2 ) 20.243 0.213 18.0802
24 B3g 2 B3g 18.9955 eV 65.27 nm 0.0000 0.2673 83.4% CV(0): B2u( 1 )-> B1u( 3 ) 20.290 0.230 18.2053
25 B2g 2 B2g 18.9955 eV 65.27 nm 0.0000 0.2673 83.4% CV(0): B3u( 1 )-> B1u( 3 ) 20.290 0.230 18.2053
26 B3u 5 B3u 19.0339 eV 65.14 nm 0.0168 1.6012 66.7% CV(1): B1u( 2 )-> B2g( 1 ) 20.612 0.715 18.2437
27 B2u 5 B2u 19.0339 eV 65.14 nm 0.0168 1.6012 66.7% CV(1): B1u( 2 )-> B3g( 1 ) 20.612 0.715 18.2437
28 Ag 4 Ag 19.0387 eV 65.12 nm 0.0000 0.0693 35.9% CO(0): Ag( 2 )-> Ag( 3 ) 21.933 0.437 18.2484
29 Ag 5 Ag 19.3341 eV 64.13 nm 0.0000 0.1694 44.7% CO(0): Ag( 2 )-> Ag( 3 ) 21.933 0.457 18.5439
30 Ag 6 Ag 19.8685 eV 62.40 nm 0.0000 1.7807 40.4% CV(1): B3u( 1 )-> B3u( 2 ) 21.084 0.338 19.0783
31 B1g 3 B1g 19.8695 eV 62.40 nm 0.0000 1.7774 40.5% CV(1): B2u( 1 )-> B3u( 2 ) 21.084 0.213 19.0792
32 B3g 3 B3g 19.9858 eV 62.04 nm 0.0000 1.6935 80.7% CV(1): B2u( 1 )-> B1u( 3 ) 21.038 0.231 19.1956
33 B2g 3 B2g 19.9858 eV 62.04 nm 0.0000 1.6935 80.7% CV(1): B3u( 1 )-> B1u( 3 ) 21.038 0.231 19.1956
34 B1g 4 B1g 19.9988 eV 62.00 nm 0.0000 1.7373 41.8% CV(1): B3u( 1 )-> B2u( 2 ) 21.084 0.213 19.2086
35 B2g 4 B2g 20.2417 eV 61.25 nm 0.0000 0.2901 81.4% CV(0): B1u( 2 )-> B3u( 2 ) 22.628 0.228 19.4515
36 B3g 4 B3g 20.2417 eV 61.25 nm 0.0000 0.2901 81.4% CV(0): B1u( 2 )-> B2u( 2 ) 22.628 0.228 19.4515
37 Au 5 Au 21.2302 eV 58.40 nm 0.0000 0.2173 40.4% CV(0): B2u( 1 )-> B2g( 2 ) 22.471 0.157 20.4400
38 B2g 5 B2g 22.1001 eV 56.10 nm 0.0000 0.0031 99.2% OV(0): Ag( 3 )-> B2g( 2 ) 23.220 0.204 21.3099
39 B3g 5 B3g 22.1001 eV 56.10 nm 0.0000 0.0031 99.2% OV(0): Ag( 3 )-> B3g( 2 ) 23.220 0.204 21.3099
40 B1g 5 B1g 23.4663 eV 52.84 nm 0.0000 0.0027 99.8% OV(0): Ag( 3 )-> B1g( 1 ) 25.135 0.283 22.6761
这里,第4、6、7激发态都是CV(1)态。注意SA-TDDFT计算的 D<S^2> 值是按U-TDDFT的公式计算出来的,可以近似地表明假如用U-TDDFT计算这些态的话,结果的自旋污染程度,但并不代表这些态实际的自旋污染程度,因为SA-TDDFT可以保证所有激发态都严格不存在自旋污染。因此如果SA-TDDFT算得的某个态的 D<S^2> 值很大,并不能表明该态的结果不可靠,相反表示对于该态而言SA-TDDFT相比U-TDDFT的改进比较大。
以闭壳层单重态为参考态计算三重态激发态
从 \(\ce{H2O}\) 分子闭壳层的基态出发,可以计算三重激发态。简洁输入为:
#! bdf.sh
tdft/b3lyp/cc-pvdz iroot=4 spinflip=1
geometry
O
H 1 R1
H 1 R1 2 109.
R1=1.0 # OH bond length in angstrom
end geometry
注意这里虽然关键词名为spinflip,但该计算并不是一个自旋翻转TDDFT计算,因为其计算的是三重态激发态的 \(M_S = 0\) 组分而非 \(M_S = 1\) 组分。对应的高级输入为:
$compass
#Notice: Coordinate unit is angstrom
geometry
O
H 1 R1
H 1 R1 2 109.
R1=1.0 # OH bond length in angstrom
end geometry
basis
cc-pvdz
group
C(1) # Force to use C1 symmetry
$end
$xuanyuan
$end
$scf
rks # ask for RKS calculation
dft
b3lyp
$end
$tddft
isf # ask for triplet TDDFT calculation
1
iroot
4
$end
TDDFT计算快结束时有输出信息如下,
*** List of excitations ***
Ground-state spatial symmetry: A
Ground-state spin: Si= 0.0000
Spin change: isf= 1
D<S^2>_pure= 2.0000 for excited state (Sf=Si+1)
D<S^2>_pure= 0.0000 for excited state (Sf=Si)
Imaginary/complex excitation energies : 0 states
Reversed sign excitation energies : 0 states
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 A 1 A 6.4131 eV 193.33 nm 0.0000 2.0000 99.2% CV(1): A( 5 )-> A( 6 ) 8.853 0.426 0.0000
2 A 2 A 8.2309 eV 150.63 nm 0.0000 2.0000 97.7% CV(1): A( 4 )-> A( 6 ) 10.736 0.519 1.8177
3 A 3 A 8.4793 eV 146.22 nm 0.0000 2.0000 98.9% CV(1): A( 5 )-> A( 7 ) 10.897 0.357 2.0661
4 A 4 A 10.1315 eV 122.37 nm 0.0000 2.0000 92.8% CV(1): A( 4 )-> A( 7 ) 12.779 0.479 3.7184
*** Ground to excited state Transition electric dipole moments (Au) ***
State X Y Z Osc.
1 0.0000 0.0000 0.0000 0.0000 0.0000
2 0.0000 0.0000 0.0000 0.0000 0.0000
3 0.0000 0.0000 0.0000 0.0000 0.0000
4 0.0000 0.0000 0.0000 0.0000 0.0000
其中, Spin change: isf= 1 提示计算的是自旋多重度比基态大2的态(也即三重态),由于基态是单重态,基态到激发态跃迁是自旋禁阻的,所以振子强度和跃迁偶极矩都是0.
TDDFT 默认只计算与参考态自旋相同的激发态, 例如,\(\ce{H2O}\) 分子的基态是单重态,TDDFT值计算单重激发态,如果要同时计算单重态与三重态,输入为:
#! H2OTDDFT.sh
TDDFT/b3lyp/cc-pVDZ iroot=4 spinflip=0,1
geometry
O
H 1 0.9
H 1 0.9 2 109.0
end geometry
系统会运行两次TDDFT,分别计算单重态和三重态,其中单重态的输出为:
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 B2 1 B2 8.0968 eV 153.13 nm 0.0292 0.0000 99.9% CV(0): B2( 1 )-> A1( 4 ) 9.705 0.415 0.0000
2 A2 1 A2 9.9625 eV 124.45 nm 0.0000 0.0000 99.9% CV(0): B2( 1 )-> B1( 2 ) 11.745 0.329 1.8656
3 A1 2 A1 10.1059 eV 122.69 nm 0.0711 0.0000 99.1% CV(0): A1( 3 )-> A1( 4 ) 11.578 0.442 2.0090
4 B1 1 B1 12.0826 eV 102.61 nm 0.0421 0.0000 99.5% CV(0): A1( 3 )-> B1( 2 ) 13.618 0.392 3.9857
5 B1 2 B1 15.1845 eV 81.65 nm 0.2475 0.0000 99.5% CV(0): B1( 1 )-> A1( 4 ) 16.602 0.519 7.0877
6 A1 3 A1 17.9209 eV 69.18 nm 0.0843 0.0000 95.4% CV(0): B1( 1 )-> B1( 2 ) 18.643 0.585 9.8240
7 A2 2 A2 22.3252 eV 55.54 nm 0.0000 0.0000 99.8% CV(0): B2( 1 )-> B1( 3 ) 24.716 0.418 14.2284
...
三重态的输出为:
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 B2 1 B2 7.4183 eV 167.13 nm 0.0000 2.0000 99.4% CV(1): B2( 1 )-> A1( 4 ) 9.705 0.415 0.0000
2 A1 1 A1 9.3311 eV 132.87 nm 0.0000 2.0000 98.9% CV(1): A1( 3 )-> A1( 4 ) 11.578 0.441 1.9128
3 A2 1 A2 9.5545 eV 129.76 nm 0.0000 2.0000 99.2% CV(1): B2( 1 )-> B1( 2 ) 11.745 0.330 2.1363
4 B1 1 B1 11.3278 eV 109.45 nm 0.0000 2.0000 97.5% CV(1): A1( 3 )-> B1( 2 ) 13.618 0.395 3.9095
5 B1 2 B1 14.0894 eV 88.00 nm 0.0000 2.0000 97.8% CV(1): B1( 1 )-> A1( 4 ) 16.602 0.520 6.6711
6 A1 2 A1 15.8648 eV 78.15 nm 0.0000 2.0000 96.8% CV(1): B1( 1 )-> B1( 2 ) 18.643 0.582 8.4465
7 A2 2 A2 21.8438 eV 56.76 nm 0.0000 2.0000 99.5% CV(1): B2( 1 )-> B1( 3 ) 24.716 0.418 14.4255
...
由于单重态到三重态跃迁是偶极禁阻的,所以振子强度 f=0.0000。对于含有重元素的体系(尤其是第三过渡周期元素),单重态到三重态可能具有一定的吸光系数,在实验上可以观测到吸收,此时需要考虑 旋轨耦合(SOC) 才能得到非零的振子强度,而且从输出文件读取振子强度的方法也与不考虑SOC的计算不同。类似地,磷光发射的振子强度在不考虑SOC时为0,只有考虑SOC才能得到非零的振子强度。
自旋翻转 (spin-flip) TDDFT计算
BDF不仅能从单重态出发计算三重态,还可以从自旋多重度更高的 2S+1 重态(S = 1/2, 1, 3/2, ...)出发,向上翻转自旋计算 2S+3 重态;自旋上翻的 TDDFT/TDA 给出的是双占据轨道的alpha电子到未占据的beta轨道跃迁态,标记为 CV(1) 激发。与基态为闭壳层单重态的情形不同,此时BDF计算的是 2S+3 重态的 \(M_S = S+1\) 组分,因此当基态不是闭壳层单重态时,该计算可以称之为自旋翻转的TDDFT计算。自旋向上翻转的TDDFT计算的输入文件格式与基态为闭壳层单重态、计算三重态激发态时完全相同,例如以下输入文件以二重态为参考态,计算四重态激发态:
...
$scf
UKS
...
spinmulti
2
$end
$tddft
...
isf
1
$end
此外,BDF还可以从三重态出发,向下翻转自旋计算单重态,这时需要设置 isf 为 -1。当然,也可以从自旋多重度更高的态向下翻转计算自旋多重度少2的态。要注意的是,自旋下翻的 TDDFT/TDA 只能正确描述从开壳层占据的alpha轨道到开壳层占据的beta轨道跃迁的电子态,标记为 OO(ab) 跃迁,其它跃迁类型的态都有自旋污染问题。
从三重态出发,向下反转自旋计算单重态,输入为:
#! H2OTDDFT.sh
TDA/b3lyp/cc-pVDZ spinmulti=3 iroot=-4 spinflip=-1
geometry
O
H 1 0.9
H 1 0.9 2 109.0
end geometry
输出为:
Imaginary/complex excitation energies : 0 states
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 A 1 A -8.6059 eV -144.07 nm 0.0000 -1.9933 99.3% OO(ab): A( 6 )-> A( 5 ) -6.123 0.408 0.0000
2 A 2 A -0.0311 eV -39809.08 nm 0.0000 -0.0034 54.1% OO(ab): A( 5 )-> A( 5 ) 7.331 1.000 8.5747
3 A 3 A 0.5166 eV 2399.85 nm 0.0000 -1.9935 54.0% OO(ab): A( 6 )-> A( 6 ) 2.712 0.999 9.1225
4 A 4 A 2.3121 eV 536.24 nm 0.0000 -0.9994 99.9% OV(ab): A( 6 )-> A( 7 ) 4.671 0.872 10.9180
这里,前三个态都是 OO(ab) 类型的激发态,其中第1个态和第3个态基本是纯的单重态(D<S^2>约等于-2,即激发态的<S^2>约等于0),第2个态基本是纯的三重态(D<S^2>约等于0);第四个态是 OV(ab) 类型的激发态,有自旋污染问题(D<S^2>约等于-1,即激发态的<S^2>约等于1,介于单重态和三重态之间),其激发能不可靠。
警告
BDF目前只支持自旋翻转的TDA,而不支持自旋翻转的TDDFT。但以闭壳层单重态为参考态计算三重态激发态不受此限制。
用iVI方法计算UV-Vis和XAS光谱
以上各算例是基于Davidson方法求解的TDDFT激发态。为了用Davidson方法求出某一个激发态,一般需要同时求解比它能量更低的所有激发态,因此当目标激发态的能量很高时(例如在计算XAS光谱时),Davidson方法需要的计算资源过多,在有限的计算时间和内存的限制下无法求得结果。此外,用户使用Davidson方法时,得到的激发态无法确保都在用户需要的波长区间内,也无法确保计算结果穷尽了用户需要的波长区间内的所有激发态。
BDF的iVI方法 [51, 52] 为以上问题提供了一种解决方案。在iVI方法中,用户可以指定感兴趣的激发能范围(比如整个可见区,或者碳的K-edge区域),而无需估计该范围内有多少个激发态;程序可以计算出激发能处于该范围内的所有激发态,一方面无需像Davidson方法那样计算比该范围的能量更低的激发态,另一方面可以确保得到该能量范围内的所有激发态,没有遗漏。以下举两个算例:
(1)计算DDQ自由基阴离子在400-700 nm范围内的吸收光谱(X-TDDFT,wB97X/LANL2DZ)
$COMPASS
Title
DDQ radical anion TDDFT
Basis
LANL2DZ
Geometry # UB3LYP/def2-SVP geometry
C 0.00000000 2.81252550 -0.25536084
C 0.00000000 1.32952185 -2.58630187
C 0.00000000 -1.32952185 -2.58630187
C 0.00000000 -2.81252550 -0.25536084
C 0.00000000 -1.29206304 2.09336443
C -0.00000000 1.29206304 2.09336443
Cl 0.00000000 -3.02272954 4.89063172
Cl -0.00000000 3.02272954 4.89063172
C 0.00000000 -2.72722649 -4.89578100
C -0.00000000 2.72722649 -4.89578100
N 0.00000000 -3.86127688 -6.78015122
N -0.00000000 3.86127688 -6.78015122
O 0.00000000 -5.15052650 -0.22779097
O -0.00000000 5.15052650 -0.22779097
End geometry
units
bohr
mpec+cosx # accelerate the calculation (both the SCF and TDDFT parts) using MPEC+COSX
$end
$XUANYUAN
rs
0.3 # rs for wB97X
$END
$SCF
roks
dft
wB97X
charge
-1
$END
$tddft
iprt # print level
2
itda
0
idiag # selects the iVI method
3
iwindow
400 700 nm # alternatively the unit can be given as au, eV or cm-1 instead of nm.
# default is in eV if no unit is given
itest
1
icorrect
1
memjkop
2048
$end
因该分子属于 \(\rm C_{2v}\) 点群,共有4个不可约表示(A1,A2,B1,B2),程序分别在4个不可约表示下求解TDDFT问题。以A1不可约表示为例,iVI迭代收敛后,程序输出如下信息:
Root 0, E= 0.1060649560, residual= 0.0002136455
Root 1, E= 0.1827715245, residual= 0.0005375061
Root 2, E= 0.1863919913, residual= 0.0006792424
Root 3, E= 0.2039707800, residual= 0.0008796108
Root 4, E= 0.2188244775, residual= 0.0015619745
Root 5, E= 0.2299349293, residual= 0.0010684879
Root 6, E= 0.2388141752, residual= 0.0618579646
Root 7, E= 0.2609321083, residual= 0.0695001907
Root 8, E= 0.2649984329, residual= 0.0759920121
Root 9, E= 0.2657352154, residual= 0.0548521587
Root 10, E= 0.2743644891, residual= 0.0655238098
Root 11, E= 0.2766959875, residual= 0.0600950472
Root 12, E= 0.2803090818, residual= 0.0587604503
Root 13, E= 0.2958382984, residual= 0.0715968457
Root 14, E= 0.3002756135, residual= 0.0607394762
Root 15, E= 0.3069930238, residual= 0.0720773993
Root 16, E= 0.3099721369, residual= 0.0956453409
Root 17, E= 0.3141986951, residual= 0.0688103843
Excitation energies of roots within the energy window (au):
0.1060649560
Timing Spin analyze : 0.01 0.00 0.00
No. 1 w= 2.8862 eV -594.3472248862 a.u. f= 0.0000 D<Pab>= 0.0717 Ova= 0.5262
CO(bb): A1( 20 )-> A2( 4 ) c_i: -0.9623 Per: 92.6% IPA: 8.586 eV Oai: 0.5360
CV(bb): A1( 20 )-> A2( 5 ) c_i: -0.1121 Per: 1.3% IPA: 11.748 eV Oai: 0.3581
CV(bb): B1( 18 )-> B2( 6 ) c_i: 0.2040 Per: 4.2% IPA: 13.866 eV Oai: 0.4328
可以看到程序在此不可约表示下计算出了17个激发态,但其中只有一个激发态(激发能0.106 au = 2.89 eV)在用户指定的波长区间(400-700 nm)内,因而完全收敛(表现为残差 (residual) 很小);其余激发态在远未收敛之前,程序即知道其不在用户感兴趣的范围内,因而不再尝试收敛这些激发态(表现为残差很大),由此节省了很多计算量。
所有4个不可约表示均计算完成后,程序照常将各不可约表示的计算结果汇总:
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 A1 2 A2 2.4184 eV 512.66 nm 0.1339 0.0280 93.0% OV(aa): A2( 4 )-> A2( 5 ) 7.064 0.781 0.0000
2 B2 1 B1 2.7725 eV 447.19 nm 0.0000 0.0000 92.5% CO(bb): B1( 18 )-> A2( 4 ) 8.394 0.543 0.3541
3 A2 1 A1 2.8862 eV 429.58 nm 0.0000 0.0000 92.6% CO(bb): A1( 20 )-> A2( 4 ) 8.586 0.526 0.4677
4 B1 1 B2 3.0126 eV 411.55 nm 0.0000 0.0000 63.5% CO(bb): B2( 4 )-> A2( 4 ) 8.195 0.820 0.5942
(2)计算乙烯的碳K-edge XAS光谱(sf-X2C,M06-2X/uncontracted def2-TZVP)
$COMPASS
Title
iVI test
Basis
def2-TZVP
geometry
C -5.77123022 1.49913343 0.00000000
H -5.23806647 0.57142851 0.00000000
H -6.84123022 1.49913343 0.00000000
C -5.09595591 2.67411072 0.00000000
H -5.62911966 3.60181564 0.00000000
H -4.02595591 2.67411072 0.00000000
End geometry
group
c(1)
uncontract # uncontract the basis set (beneficial for the accuracy of core excitations)
$END
$XUANYUAN
heff
3 # selects sf-X2C
$END
$SCF
rks
dft
m062x
$END
$TDDFT
imethod
1 # R-TDDFT
idiag
3 # iVI
iwindow
275 285 # default unit: eV
$end
由实验得知碳的K-edge吸收在280 eV附近,因此这里的能量范围选为275-285 eV。计算得到该能量区间内共有15个激发态:
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 A 2 A 277.1304 eV 4.47 nm 0.0018 0.0000 97.1% CV(0): A( 5 )-> A( 93 ) 281.033 0.650 0.0000
2 A 3 A 277.1998 eV 4.47 nm 0.0002 0.0000 96.0% CV(0): A( 6 )-> A( 94 ) 282.498 0.541 0.0694
3 A 4 A 277.9273 eV 4.46 nm 0.0045 0.0000 92.8% CV(0): A( 7 )-> A( 94 ) 281.169 0.701 0.7969
4 A 5 A 278.2593 eV 4.46 nm 0.0000 0.0000 100.0% CV(0): A( 8 )-> A( 95 ) 283.154 0.250 1.1289
5 A 6 A 279.2552 eV 4.44 nm 0.0002 0.0000 85.5% CV(0): A( 4 )-> A( 93 ) 284.265 0.627 2.1247
6 A 7 A 280.0107 eV 4.43 nm 0.0000 0.0000 96.6% CV(0): A( 8 )-> A( 96 ) 284.941 0.315 2.8803
7 A 8 A 280.5671 eV 4.42 nm 0.0000 0.0000 97.0% CV(0): A( 5 )-> A( 94 ) 284.433 0.642 3.4366
8 A 9 A 280.8642 eV 4.41 nm 0.1133 0.0000 93.3% CV(0): A( 2 )-> A( 9 ) 287.856 0.179 3.7337
9 A 10 A 280.8973 eV 4.41 nm 0.0000 0.0000 90.1% CV(0): A( 1 )-> A( 9 ) 287.884 0.185 3.7668
10 A 11 A 281.0807 eV 4.41 nm 0.0000 0.0000 66.8% CV(0): A( 6 )-> A( 95 ) 287.143 0.564 3.9502
11 A 12 A 282.6241 eV 4.39 nm 0.0000 0.0000 97.7% CV(0): A( 7 )-> A( 95 ) 285.815 0.709 5.4937
12 A 13 A 283.7528 eV 4.37 nm 0.0000 0.0000 65.1% CV(0): A( 4 )-> A( 94 ) 287.666 0.592 6.6223
13 A 14 A 283.9776 eV 4.37 nm 0.0000 0.0000 92.1% CV(0): A( 6 )-> A( 96 ) 288.929 0.523 6.8471
14 A 15 A 284.1224 eV 4.36 nm 0.0008 0.0000 98.2% CV(0): A( 7 )-> A( 96 ) 287.601 0.707 6.9920
15 A 16 A 284.4174 eV 4.36 nm 0.0000 0.0000 93.7% CV(0): A( 3 )-> A( 93 ) 289.434 0.509 7.2869
但由激发态成分可以看出,只有激发能为280.8642 eV和280.8973 eV的两个激发态为C 1s到价层轨道的激发,其余激发均为价层轨道到非常高的Rydberg轨道的激发,也即对应于价层电子电离的背景吸收。
此外,即使用户没有不重不漏地计算某能量区间内所有激发态的需求,而是仅需要计算给定数量的激发态,iVI相比Davidson方法仍有另一优势,即在该情况下所需内存较少。Davidson方法所需内存随迭代次数的增加而线性增加,虽然BDF采取分批计算激发态、以及每几十步迭代重新构建Krylov子空间的方法减少内存消耗,但这会导致迭代次数增加,从而增加计算时间。而iVI方法因为每步迭代时都重新构建Krylov子空间,算法消耗的内存并不随迭代的进行而增加,相比Davidson方法可以节省2~10倍的内存消耗。因此,当Davidson方法所需内存超过当前节点的可用物理内存,但超过的比例在10倍以内时,改用iVI方法有一定概率可以让计算在给定的内存限制下正常完成。例如以下写法
$TDDFT
idiag
3 # iVI
iroot
-100
$end
即为用iVI方法计算能量最低的100个自旋守恒激发态。当内存足够时,计算时间与Davidson方法相差不多;当内存不能满足Davidson方法的需求,但差距不太远时,Davidson方法会因内存不足而报错退出,或因频繁重新构建Krylov子空间而导致迭代次数变多(甚至不收敛),而iVI方法仍可正常收敛。不过,若用户以iwindow关键字指定需要计算的波长/激发能范围,而非用iroot、Nroot指定需要计算的激发态数目,则iVI所需的内存可能多于Davidson方法所需内存,因为iVI需要消耗额外内存用以确保没有遗漏iwindow之内的任何激发态。
快速近似计算大体系吸收光谱的方法:sTDA、sTDDFT
传统的TDDFT方法在计算大体系(例如数百个原子)的吸收光谱时,常常会遇到严重的CPU和内存瓶颈,导致计算无法在给定的计算时间和内存限制下完成。这不仅是因为计算每个激发态所需的计算资源变多,更是因为体系越大,其在一定波长范围(如可见光区)内的激发态数目就越多。因此,如果要计算某给定波长范围内的吸收光谱,则TDDFT计算所需时间和内存消耗不仅随体系大小快速增加,其与SCF步骤所需时间和内存的比值也是随体系大小的增加而增加的。也就是说,当体系足够大时,即使只对TDDFT步骤做近似,而不对SCF步骤做近似,也可以获得极大的加速,并节省大量内存。如上所述,iVI方法可以在一定程度上减少TDDFT计算所需内存,且不引入任何误差;而 MPEC+COSX方法 则可将TDDFT的计算时间降低至原来的1/10~1/3左右(视基组大小和体系大小而定),代价是引入极小(一般小于0.01 eV)的误差。但如果对结果精度的要求更低,例如即使0.2 eV量级的误差也可以接受,则可以利用Grimme课题组发展的sTDA、sTDDFT方法 [70, 71, 72] 加速TDDFT计算,可较普通TDDFT快数十至数百倍。在BDF里,可用 grimmestd 关键词来指定使用sTDA或sTDDFT方法。
例如,以下算例用sTDDFT计算叶绿素a(137个原子)的吸收光谱:
$compass
title
chlorophyll a
basis
def2-sv(p)
geometry
Mg -6.39280500 1.01913900 0.07930600
C -4.66061700 -1.97549200 0.32240100
C -3.86800400 2.56481900 1.82052600
C -8.08215800 3.98978800 -0.18167200
C -8.98545300 -0.61768600 -1.64547000
N -4.54433200 0.38436500 0.90884900
C -3.99700200 -0.93553500 0.86684800
C -3.70478200 1.19580500 1.58959100
N -6.02943300 2.90039700 0.68978700
C -4.94074100 3.33410600 1.39121000
C -5.07491500 4.81749500 1.63863600
C -6.24086300 5.22118200 1.06806800
C -6.89203100 4.01489100 0.45469200
C -4.06725100 5.61005500 2.36565900
C -6.80943200 6.56357900 1.03550500
C -7.16536900 7.19003700 -0.08627800
N -8.20213100 1.58193300 -0.75743000
C -8.71213700 2.83175300 -0.76290000
C -10.01431500 2.85490100 -1.44851000
C -10.27039900 1.56409200 -1.85400400
C -9.13329500 0.73615200 -1.42942600
C -10.84075600 4.06541800 -1.63406700
N -6.79660200 -0.84366300 -0.52933900
C -7.89913200 -1.40200500 -1.24381700
C -7.66635200 -2.82277100 -1.44961100
C -6.43617900 -3.10668000 -0.86460900
C -5.95222300 -1.85130000 -0.31154100
C -8.56834600 -3.75605800 -2.14493700
C -5.45761400 -4.14091100 -0.60755600
O -5.41067600 -5.29722700 -0.93531800
C -4.27700300 -3.43898300 0.19681800
C -4.03436300 -4.04185800 1.55541600
O -2.98821400 -4.06496400 2.17129100
O -5.18821800 -4.55887600 2.07822700
C -5.09043500 -5.21072200 3.37451000
H -3.08326400 3.06907300 2.38501100
H -8.64877900 4.92413800 -0.27855400
H -9.79244500 -1.13563000 -2.18571200
H -3.93018000 5.23884000 3.39358500
H -3.08555400 5.56125900 1.86717500
H -4.34148300 6.67290700 2.43393200
H -6.91464100 7.03432600 2.01872100
H -7.57843000 8.18875500 -0.09998800
H -7.06020700 6.75751400 -1.07293700
H -8.14333300 -4.77543300 -2.17957800
H -8.75310000 -3.45058300 -3.18537500
H -9.54347000 -3.83344900 -1.64123300
H -6.14095000 -5.40216500 3.61932300
H -4.61251400 -4.54263500 4.09691600
H -4.52176200 -6.13925800 3.26271900
H -11.76604400 3.85006500 -2.18728300
H -10.29928900 4.83683900 -2.20105400
H -11.13298700 4.50356100 -0.66841600
H -3.34289100 -3.55371300 -0.41277200
C -11.45722200 1.05206800 -2.59092400
H -11.76806300 0.06727900 -2.18361200
H -12.32721500 1.72374600 -2.42522700
C -11.17530300 0.93618900 -4.08970000
H -10.32963900 0.26795200 -4.29109700
H -12.04576500 0.54981100 -4.62999500
H -10.91967800 1.91226500 -4.52115700
C -2.62887700 -0.98246300 1.53480600
H -2.66523600 -1.73547400 2.36545400
C -2.45989500 0.45470900 2.10966600
H -1.54474300 0.93905400 1.69345300
C -1.51912600 -1.36887400 0.54488500
H -1.95440500 -1.82032400 -0.37473000
H -0.98048400 -0.46992100 0.18497700
C -0.53490800 -2.35906300 1.17264300
H -0.01435300 -1.91575300 2.04669100
H -1.09048500 -3.24472000 1.58712500
C 0.45366200 -2.85133200 0.15756500
O 0.32298700 -3.00078100 -1.03465600
O 1.62455500 -3.17223400 0.80990800
C 2.74348900 -3.67458400 0.01127500
H 3.16253400 -4.45724900 0.67208000
H 2.35407200 -4.12003600 -0.92533200
C -2.39399700 0.47145400 3.63155500
H -1.53316200 -0.10264900 3.99668600
H -2.29784400 1.49298200 4.01962300
H -3.29480800 0.03786900 4.08539800
C 3.69329800 -2.54884800 -0.22275100
H 3.47934900 -1.65803400 0.36902200
C 4.72857100 -2.60301500 -1.07403300
C 5.65017100 -1.42380300 -1.25339300
H 5.14884400 -0.48370900 -0.94555600
H 5.88443700 -1.28751700 -2.32864900
C 5.03510200 -3.81649000 -1.89435600
H 5.11655600 -4.71792300 -1.27224100
H 4.24043400 -3.99998600 -2.63355100
H 5.97637900 -3.72648800 -2.45109500
C 6.94460300 -1.61032500 -0.44635600
H 6.69651300 -1.73292300 0.62680900
H 7.44457000 -2.55070000 -0.74876300
C 7.89779300 -0.42393400 -0.63427700
H 7.40043300 0.51456700 -0.32490500
H 8.12487300 -0.30133700 -1.71103300
C 9.21414800 -0.60223000 0.15481900
H 9.61685800 -1.62347600 -0.05750700
C 8.97090200 -0.48135200 1.66411800
H 8.57313200 0.50305400 1.93258400
H 8.25269000 -1.23110800 2.01368400
H 9.89846400 -0.62443600 2.22911700
C 10.24945900 0.43890900 -0.32513700
H 10.24713000 0.48183100 -1.43148900
H 9.95072700 1.44860700 0.01380100
C 11.66689200 0.11913500 0.16783800
H 11.68178700 0.08831400 1.27533400
H 11.96235100 -0.89412300 -0.16596100
C 12.68264200 1.15206500 -0.33770400
H 12.39293700 2.16426800 0.00143900
H 12.65111300 1.18669100 -1.44390400
C 14.12108800 0.83574000 0.12861700
H 14.33172200 -0.24146100 -0.08434100
C 14.27459700 1.07059200 1.63652100
H 13.57809500 0.44876700 2.20914700
H 15.28809800 0.82990700 1.97526900
H 14.07897900 2.11411800 1.90509100
C 15.12505600 1.69543200 -0.67097600
H 14.85566900 1.67748900 -1.74474600
H 15.04336200 2.75380800 -0.36005400
C 16.57081500 1.21005300 -0.50195300
H 16.85440700 1.23936500 0.56866100
H 16.64949400 0.14854000 -0.80588000
C 17.54788100 2.06201800 -1.32247100
H 17.47406000 3.12251900 -1.01707800
H 17.25297400 2.03835700 -2.38919200
C 19.00728700 1.57806500 -1.18264700
H 19.02932300 0.46921900 -1.32861700
C 19.88192000 2.22132000 -2.26846200
H 19.87986900 3.31392300 -2.19414300
H 20.92289700 1.89145300 -2.18575500
H 19.53365000 1.95811100 -3.27242200
C 19.57038500 1.89281000 0.20940000
H 19.59163600 2.97072900 0.40174700
H 18.96496600 1.43221300 0.99745100
H 20.59391000 1.51998700 0.31823800
end geometry
$end
$xuanyuan
$end
$scf
rks
dft
b3lyp
$end
$tddft
iwindow
300 700 nm
grimmestd
$end
计算的SCF部分耗时527 s(16线程OpenMP并行,下同),TDDFT部分耗时仅152 s,得到以下的激发能和振子强度信息:
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 A 2 A 2.1820 eV 568.22 nm 0.2526 0.0000 75.2% CV(0): A( 241 )-> A( 242 ) 2.473 0.725 0.0000
2 A 3 A 2.3886 eV 519.07 nm 0.0141 0.0000 60.8% CV(0): A( 240 )-> A( 242 ) 2.922 0.731 0.2066
3 A 4 A 3.0363 eV 408.34 nm 0.0101 0.0000 88.5% CV(0): A( 237 )-> A( 242 ) 3.896 0.368 0.8544
4 A 5 A 3.1122 eV 398.38 nm 0.0190 0.0000 92.1% CV(0): A( 239 )-> A( 242 ) 3.725 0.498 0.9302
5 A 6 A 3.1769 eV 390.27 nm 0.4325 0.0000 36.3% CV(0): A( 241 )-> A( 243 ) 3.179 0.662 0.9949
6 A 7 A 3.2453 eV 382.04 nm 0.0516 0.0000 86.5% CV(0): A( 236 )-> A( 242 ) 3.931 0.542 1.0634
7 A 8 A 3.2665 eV 379.57 nm 0.0007 0.0000 98.9% CV(0): A( 238 )-> A( 242 ) 3.748 0.030 1.0845
8 A 9 A 3.4194 eV 362.59 nm 0.6594 0.0000 50.2% CV(0): A( 240 )-> A( 243 ) 3.628 0.649 1.2375
9 A 10 A 3.5309 eV 351.14 nm 0.4136 0.0000 76.8% CV(0): A( 235 )-> A( 242 ) 4.125 0.577 1.3489
10 A 11 A 3.7388 eV 331.62 nm 0.0348 0.0000 93.3% CV(0): A( 239 )-> A( 243 ) 4.430 0.544 1.5568
11 A 12 A 3.7606 eV 329.69 nm 0.0599 0.0000 83.4% CV(0): A( 241 )-> A( 244 ) 4.229 0.648 1.5786
12 A 13 A 3.8813 eV 319.44 nm 0.0033 0.0000 94.2% CV(0): A( 237 )-> A( 243 ) 4.601 0.269 1.6993
13 A 14 A 3.9358 eV 315.01 nm 0.1686 0.0000 67.2% CV(0): A( 234 )-> A( 242 ) 4.532 0.633 1.7539
14 A 15 A 3.9750 eV 311.91 nm 0.0000 0.0000 99.7% CV(0): A( 238 )-> A( 243 ) 4.453 0.028 1.7930
15 A 16 A 4.0250 eV 308.04 nm 0.0187 0.0000 56.9% CV(0): A( 236 )-> A( 243 ) 4.636 0.512 1.8430
16 A 17 A 4.0346 eV 307.30 nm 0.0697 0.0000 32.9% CV(0): A( 233 )-> A( 242 ) 4.697 0.464 1.8526
17 A 18 A 4.0803 eV 303.86 nm 0.0461 0.0000 57.5% CV(0): A( 241 )-> A( 245 ) 4.702 0.492 1.8983
18 A 19 A 4.1011 eV 302.32 nm 0.0046 0.0000 49.1% CV(0): A( 233 )-> A( 242 ) 4.697 0.418 1.9192
相比之下,传统的TDDFT计算(与以上输入文件相同,区别仅在于去掉 grimmestd 关键字)耗时3264 s,结果为:
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 A 2 A 2.2098 eV 561.08 nm 0.2224 0.0000 77.3% CV(0): A( 241 )-> A( 242 ) 2.473 0.724 0.0000
2 A 3 A 2.4379 eV 508.56 nm 0.0085 0.0000 60.0% CV(0): A( 240 )-> A( 242 ) 2.922 0.733 0.2282
3 A 4 A 3.1690 eV 391.24 nm 0.1398 0.0000 35.3% CV(0): A( 239 )-> A( 242 ) 3.725 0.490 0.9592
4 A 5 A 3.1923 eV 388.39 nm 0.0011 0.0000 49.7% CV(0): A( 239 )-> A( 242 ) 3.725 0.428 0.9825
5 A 6 A 3.2259 eV 384.34 nm 0.3826 0.0000 31.2% CV(0): A( 241 )-> A( 243 ) 3.179 0.608 1.0161
6 A 7 A 3.3241 eV 372.99 nm 0.0528 0.0000 88.4% CV(0): A( 236 )-> A( 242 ) 3.931 0.547 1.1143
7 A 8 A 3.4675 eV 357.56 nm 0.7779 0.0000 67.6% CV(0): A( 240 )-> A( 243 ) 3.628 0.667 1.2577
8 A 9 A 3.5022 eV 354.02 nm 0.0052 0.0000 99.4% CV(0): A( 238 )-> A( 242 ) 3.748 0.028 1.2925
9 A 10 A 3.5947 eV 344.91 nm 0.2244 0.0000 89.5% CV(0): A( 235 )-> A( 242 ) 4.125 0.561 1.3849
10 A 11 A 3.7945 eV 326.75 nm 0.0343 0.0000 88.7% CV(0): A( 239 )-> A( 243 ) 4.430 0.550 1.5847
11 A 12 A 3.8277 eV 323.92 nm 0.0463 0.0000 84.3% CV(0): A( 241 )-> A( 244 ) 4.229 0.648 1.6179
12 A 13 A 4.0449 eV 306.52 nm 0.0860 0.0000 72.5% CV(0): A( 234 )-> A( 242 ) 4.532 0.644 1.8351
13 A 14 A 4.0913 eV 303.04 nm 0.0021 0.0000 95.9% CV(0): A( 237 )-> A( 243 ) 4.601 0.264 1.8815
可以看到两个计算的激发能差别极小,在0.0~0.2 eV量级。表面上看,有一些态的振子强度差别很大,但这是因为激发能非常接近的态彼此之间混合的结果,如果作出光谱图(见 高斯展宽的吸收光谱的绘制 ),可以发现sTDDFT和TDDFT的吸收光谱基本相同,两者的差别在DFT计算的正常误差范围内:
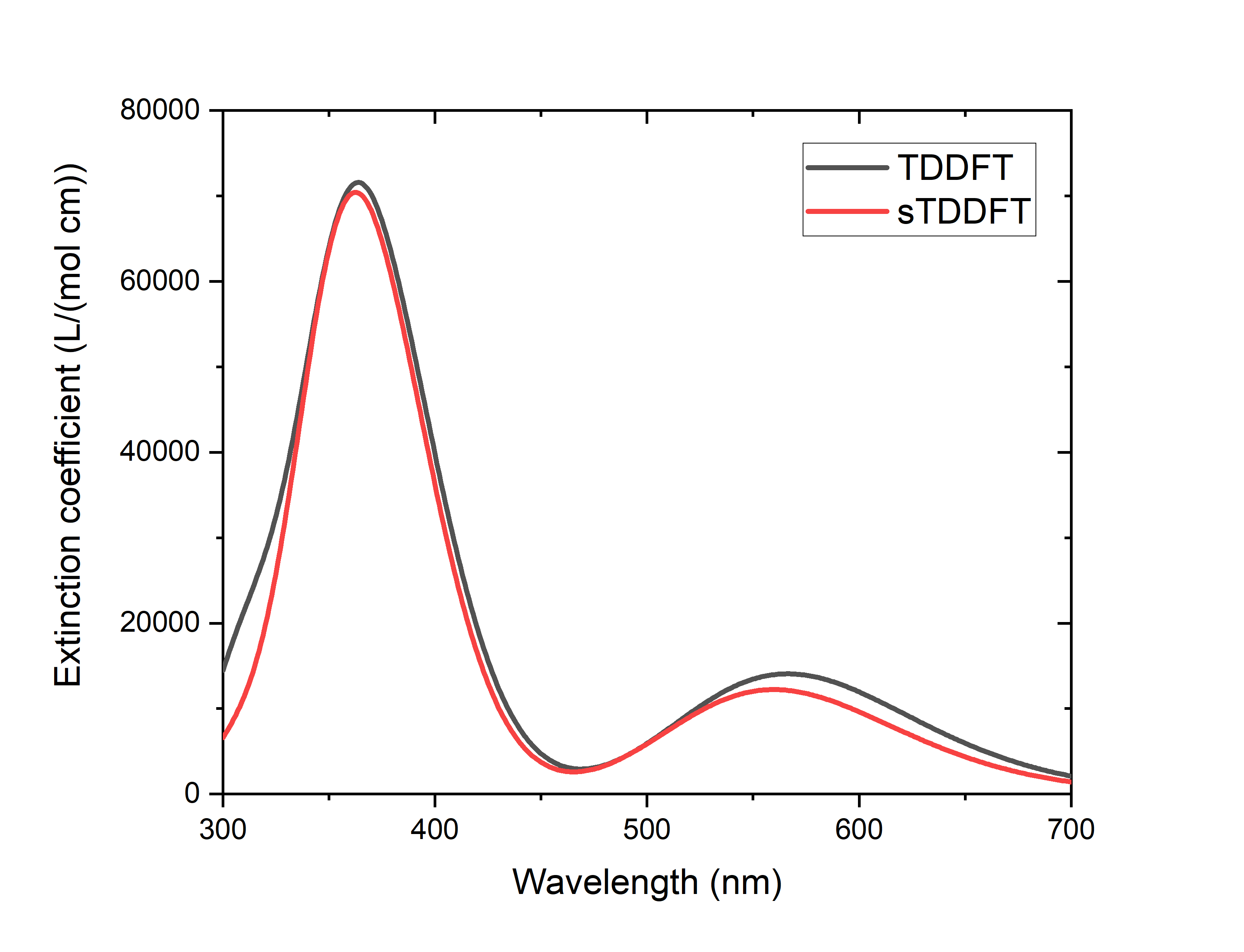
与此同时,sTDDFT相比TDDFT节省了95 %的TDDFT计算时间(如包括SCF的计算时间,则节省了84 %的计算时间),可见加速效果十分可观。
除sTDDFT外,还可以将 grimmestd 关键字用于TDA计算,来指定进行sTDA计算,例如:
$tddft
itda
1
iwindow
300 700 nm
grimmestd
$end
当然,也可以指定计算的激发态数而非波长范围:
$tddft
nroot # calculate 100 lowest excited states per irrep
100
grimmestd
$end
更多注意事项请参见 grimmestd关键字的介绍 。
重启被意外中断的TDDFT任务
如TDDFT计算被意外终止,用户可能希望进行断点续算,即在重新做TDDFT计算的时候,利用之前的被中断的TDDFT任务产生的一些中间结果,来减少或避免重复计算。关于TDDFT计算断点续算的方法,详见 “常见问题”一章里的相应介绍 。
高斯展宽的吸收光谱的绘制
以上各计算得到的仅是各个激发态的激发能和振子强度,而用户常常需要得到理论预测的吸收谱的峰形,这就需要把每个激发态的吸收按一定的半峰宽进行高斯展宽。在BDF中,这是通过Python脚本plotspec.py(位于$BDFHOME/sbin/下,其中$BDFHOME是BDF的安装路径)来实现的。用户需要在TDDFT计算完成以后,手动从命令行调用plotspec.py。例如假设我们已经用BDF计算得到了C60分子的TDDFT激发态,对应的输出文件为C60.out,则可以运行
$BDFHOME/sbin/plotspec.py C60.out
或者
$BDFHOME/sbin/plotspec.py C60
该脚本会在屏幕上输出以下信息:
==================================
P L O T S P E C
Spectral broadening tool for BDF
==================================
BDF output file: C60.out
1 TDDFT output block(s) found
Block 1: 10 excited state(s)
- Singlet absorption spectrum, spin-allowed
The spectra will be Gaussian-broadened (FWHM = 0.5000 eV) ...
Absorption maxima of spectrum 1 (nm (lg epsilon/(L/(mol cm)))):
- 238 (5.12), 308 (4.50)
plotspec.py: exit successfully
并产生两个文件,一个是C60.stick.csv,包含所有激发态的吸收波长和摩尔消光系数,可以用来作棒状图:
TDDFT Singlets 1,,
Wavelength,Extinction coefficient,
nm,L/(mol cm),
342.867139,2899.779319,
307.302300,31192.802393,
237.635960,131840.430395,
211.765024,295.895849,
209.090150,134.498113,
197.019205,179194.526059,
178.561512,145.257962,
176.943322,54837.570677,
164.778366,548.752301,
160.167663,780.089056,
另一个是C60.spec.csv,包含高斯展宽后的吸收谱(默认的展宽FWHM为0.5 eV):
TDDFT Singlets 1,,
Wavelength,Extinction coefficient,
nm,L/(mol cm),
200.000000,162720.545118,
201.000000,151036.824457,
202.000000,137429.257570,
...
998.000000,0.000000,
999.000000,0.000000,
1000.000000,0.000000,
这两个文件可以用Excel、Origin等作图软件打开并作图:
可以用命令行参数控制作图范围、高斯展宽的FWHM等。示例:
# Plot the spectrum in the range 300-600 nm:
$BDFHOME/sbin/plotspec.py wavelength=300-600nm filename.out
# Plot an X-ray absorption spectrum in the range 200-210 eV,
# using an FWHM of 1 eV:
$BDFHOME/sbin/plotspec.py energy=200-210eV fwhm=1eV filename.out
# Plot a UV-Vis spectrum in the range 10000 cm-1 to 40000 cm-1,
# where the wavenumber is sampled at an interval of 50 cm-1:
$BDFHOME/sbin/plotspec.py wavenumber=10000-40000cm-1 interval=50 filename.out
# Plot an emission spectrum in the range 600-1200 nm, as would be
# given by Kasha's rule (i.e. only the first excited state is considered),
# where the wavelength is sampled at an interval of 5 nm:
$BDFHOME/sbin/plotspec.py -emi wavelength=600-1200nm interval=5 filename.out
如果不带命令行参数运行$BDFHOME/sbin/plotspec.py,可以列出所有的命令行参数及用法,这里不予赘述。
电子圆二色性(ECD)谱的计算
除吸收光谱外,BDF还支持在TDDFT级别下计算圆二色性(ECD)谱。用户只需在$tddft模块的输入中添加ECD关键字即可。例如以下输入文件在wB97X/ma-def2-TZVP水平下计算(S)-5-甲基环戊-2-烯-1-酮在160-300 nm范围内的ECD谱,溶剂为水:
$COMPASS
Title
ECD test
Basis
ma-def2-TZVP
Geometry # B3LYP/def2-SVP geometry
C 11.03017501307698 -1.06358915357097 18.65132535474617
C 12.57384005718525 -1.02456284484694 18.65658561738920
C 12.91529117412091 0.43177145174825 18.82255138315294
C 11.83078974644673 1.23189442235475 18.82242608164620
H 10.67388955940226 -1.47007769437446 19.61628109972719
H 13.00096293117676 -1.40629079282790 17.71067917782706
H 13.02306939327327 -1.63533989080155 19.45869631125239
H 13.94838829748073 0.77963695466942 18.91842719115154
H 11.81586135485978 2.32060314334658 18.90537981712256
C 10.61010494985639 0.41685642111484 18.65633627754937
O 9.46516754355473 0.82239910074197 18.54006339565965
C 10.37591484801120 -1.85714650215417 17.51891751829459
H 10.61141701850992 -2.93014535161767 17.59810807151853
H 9.28153845878811 -1.73962079399751 17.55678289237466
H 10.72376849425688 -1.50217177978463 16.53426564058783
End Geometry
MPEC+COSX
$END
$XUANYUAN
rs
0.3
$END
$SCF
RKS
DFT
wB97X
solvent
water
solmodel
SMD # IEFPCM is also a reasonable choice and is almost equally accurate
$END
$tddft
iprt
3
# To ensure that we get all roots within the window, we use the iVI method.
# Nevertheless, of course, iVI is not mandatory for ECD calculations
idiag
3 # use iVI
iwindow
160 300 nm
ecd # specifies ECD calculation
solneqlr # linear response solvation, recommended when the number of excited states
# is large
$end
该计算在输出吸收波长、振子强度、跃迁偶极矩后,还输出了跃迁磁偶极矩,以及长度、速度表象下的转子强度:
*** Ground to excited state Transition magnetic dipole moments (Au) ***
State X Y Z
1 -0.001936 0.002882 0.000034
2 -0.000444 -0.000188 -0.004692
3 -0.000342 -0.003475 -0.000070
4 -0.001232 0.000479 -0.001992
5 0.000581 0.002272 -0.001047
6 -0.001917 0.003593 -0.000178
7 0.002065 0.000206 -0.000823
*** Electronic circular dichroism (ECD) rotatory strengths (1e-40 cgs) ***
State Length formalism Velocity formalism
1 -2.9144 -3.0791
2 18.0007 17.5760
3 -25.1038 -25.1132
4 -7.2316 -7.0551
5 25.1323 24.4034
6 -14.9753 -14.2051
7 -30.6305 -30.8057
接下来,可以用plotspec.py读取该输出文件中的转子强度信息,进行高斯展宽,得到.spec.csv和.stick.csv文件,作ECD图(其中因为吸收波长略小于160 nm的激发态在高斯展宽后会对160 nm附近的图谱有一定影响,160 nm附近的ECD谱可能不可靠,因此作图时只作到180 nm):
$BDFHOME/sbin/plotspec.py -cd wavelength=180-300 filename.out
结果如下:
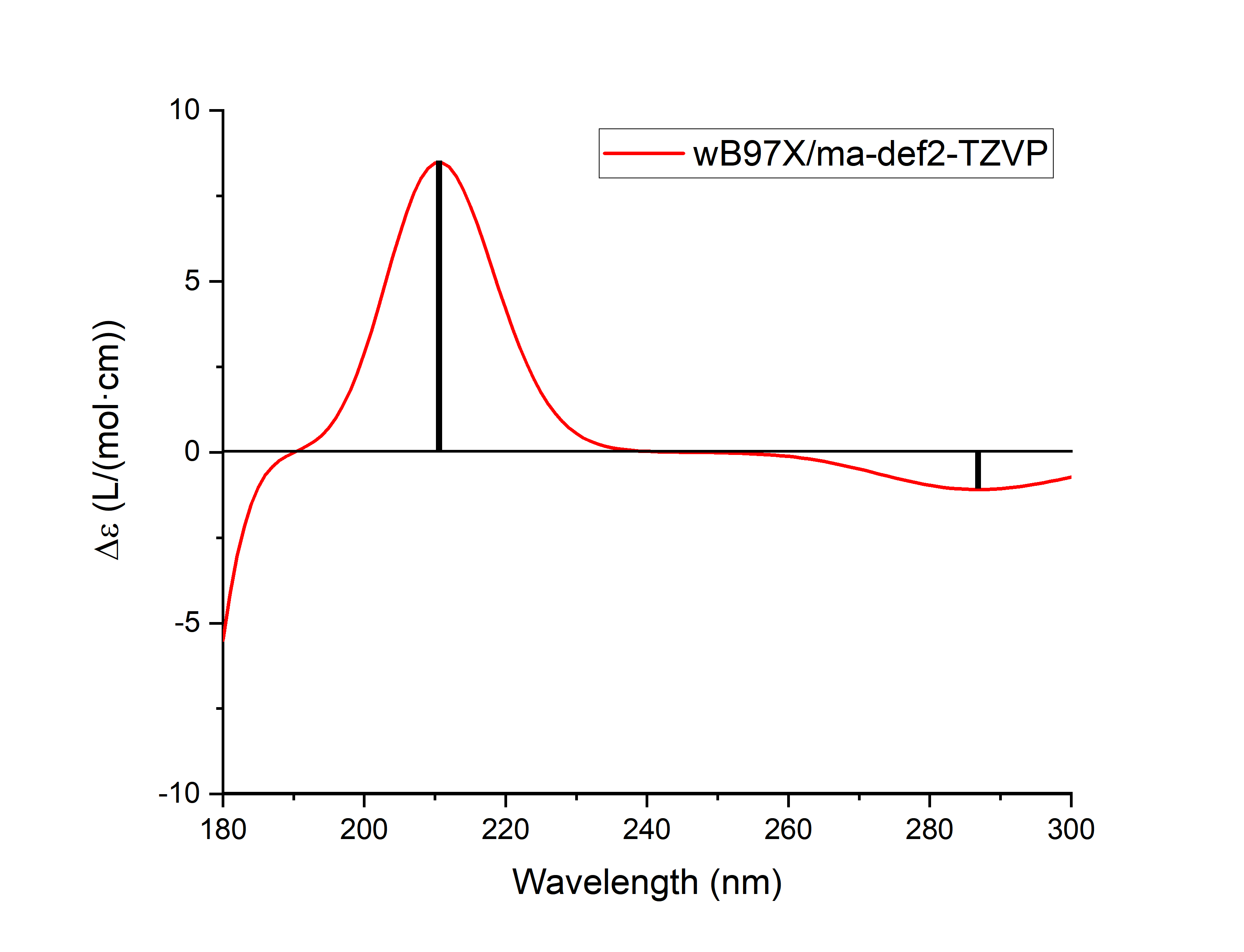
备注
虽然当基组趋于完备时,速度表象下的转子强度和长度表象下的转子强度严格相等,但当基组大小有限时,速度表象下的转子强度不依赖于分子的取向和中心位置,但长度表象下的转子强度是依赖于分子的取向和中心位置的。因此,起码当分子比较大、基组不太大的情况下,速度表象下的转子强度结果较为可靠。plotspec.py默认是用速度表象下的转子强度来绘制ECD图的,如需要用长度表象下的转子强度来绘制ECD图,应将plotspec.py的命令行参数中的-cd改为-cdl。
因BDF和其他程序确定分子标准取向以及分子坐标原点的方法不同,BDF计算出的长度表象下的转子强度可能与其他程序存在少许差别,这是正常现象,是由上述长度表象下的转子强度的理论缺陷所导致的,不代表计算结果错误。但速度表象下的转子强度理应和其他程序吻合较好。
对于柔性分子,单一构象的ECD计算结果不可靠,建议结合CREST、Molclus等软件进行构象搜索,对所有主要构象分别计算ECD谱后,进行Boltzmann加权平均。
激发态结构优化
BDF不仅支持TDDFT单点能(即给定分子结构下的激发能)的计算,还支持激发态的结构优化、数值频率等计算。为此需要在 $tddft 模块之后添加 $resp 模块用于计算TDDFT能量的梯度,并在 $compass 模块后添加 $bdfopt 模块,利用TDDFT梯度信息进行结构优化和频率计算(详见 结构优化与频率计算 )。
以下是在B3LYP/cc-pVDZ水平下优化丁二烯第一激发态结构的算例:
$COMPASS
Title
C4H6
Basis
CC-PVDZ
Geometry # Coordinates in Angstrom. The structure has C(2h) symmetry
C -1.85874726 -0.13257980 0.00000000
H -1.95342119 -1.19838319 0.00000000
H -2.73563916 0.48057645 0.00000000
C -0.63203020 0.44338226 0.00000000
H -0.53735627 1.50918564 0.00000000
C 0.63203020 -0.44338226 0.00000000
H 0.53735627 -1.50918564 0.00000000
C 1.85874726 0.13257980 0.00000000
H 1.95342119 1.19838319 0.00000000
H 2.73563916 -0.48057645 0.00000000
End Geometry
$END
$BDFOPT
solver
1
$END
$XUANYUAN
$END
$SCF
RKS
dft
B3lyp
$END
$TDDFT
nroot
# The ordering of irreps of the C(2h) group is: Ag, Au, Bg, Bu
# Thus the following line specifies the calculation of the 1Bu state, which
# happens to be the first excited state for this particular molecule.
0 0 0 1
istore
1
# TDDFT gradient requires tighter TDDFT convergence criteria than single-point
# TDDFT calculations, thus we tighten the convergence criteria below.
crit_vec
1.d-6 # default 1.d-5
crit_e
1.d-8 # default 1.d-7
$END
$resp
geom
norder
1 # first-order nuclear derivative
method
2 # TDDFT response properties
nfiles
1 # must be the same number as the number after the istore keyword in $TDDFT
iroot
1 # calculate the gradient of the first root. Can be omitted here since only
# one root is calculated in the $TDDFT block
$end
注意上述算例中, $resp 模块的关键词 iroot 的意义和前述 $tddft 模块的关键词 iroot 的意义不同。前者指的是计算第几个激发态的梯度,后者则指的是每个不可约表示一共计算多少个激发态。
结构优化收敛后,在主输出文件中输出收敛的结构:
Good Job, Geometry Optimization converged in 5 iterations!
Molecular Cartesian Coordinates (X,Y,Z) in Angstrom :
C -1.92180514 0.07448476 0.00000000
H -2.21141426 -0.98128927 0.00000000
H -2.70870517 0.83126705 0.00000000
C -0.54269837 0.45145649 0.00000000
H -0.31040658 1.52367715 0.00000000
C 0.54269837 -0.45145649 0.00000000
H 0.31040658 -1.52367715 0.00000000
C 1.92180514 -0.07448476 0.00000000
H 2.21141426 0.98128927 0.00000000
H 2.70870517 -0.83126705 0.00000000
Force-RMS Force-Max Step-RMS Step-Max
Conv. tolerance : 0.2000E-03 0.3000E-03 0.8000E-03 0.1200E-02
Current values : 0.5550E-04 0.1545E-03 0.3473E-03 0.1127E-02
Geom. converge : Yes Yes Yes Yes
此外可以从 .out.tmp 文件的最后一个TDDFT模块的输出里读取激发态平衡结构下的激发能,以及激发态的总能量、主要成分:
No. 1 w= 5.1695 eV -155.6874121542 a.u. f= 0.6576 D<Pab>= 0.0000 Ova= 0.8744
CV(0): Ag( 6 )-> Bu( 10 ) c_i: 0.1224 Per: 1.5% IPA: 17.551 eV Oai: 0.6168
CV(0): Bg( 1 )-> Au( 2 ) c_i: -0.9479 Per: 89.9% IPA: 4.574 eV Oai: 0.9035
...
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 Bu 1 Bu 5.1695 eV 239.84 nm 0.6576 0.0000 89.9% CV(0): Bg( 1 )-> Au( 2 ) 4.574 0.874 0.0000
其中,激发态平衡结构下的激发能对应的波长(240 nm)即为丁二烯的荧光发射波长。
备注
某些体系的激发态结构优化会振荡不收敛,这一般是因为优化到了锥形交叉点附近;如果优化到了激发态和基态的锥形交叉点附近,且用的是Full TDDFT而非TDA,则结构优化甚至可能会因激发能变为虚数或复数而报错退出。这两种情况是正常现象,其成因及解决方案详见 几何优化不收敛的解决方法 。
在以上的算例中,我们跟踪的是绝热态。自2025年11月起,BDF支持在结构优化中跟踪透热态,即根据激发态成分而非激发态的序号,来确定结构优化的下一步应该跟踪哪个态、算哪个态的梯度。方法是将 $resp 模块的 iroot 值设为负,如
$resp
...
iroot
-2
$end
表示:结构优化的第一步跟踪第2个激发态,之后第N+1步(N=1,2,...)跟踪和第N步跟踪的激发态成分最相似的态。当然这也要求 $tddft 模块计算的激发态数大于等于2,且最好大于2。
在结构优化的第二步,可以看到 $resp 模块有类似下面的输出
Root 1 overlap = .97267
Root 2 overlap = .85203E-02
Root 3 overlap = .84548E-03
Root 4 overlap = .10493E-02
Best match: Root 1 of the previous geometry step, overlap 0.97267
Second best match: Root 2 of the previous geometry step, overlap 0.00852
Will follow Root 1
在本例中,程序计算得到当前结构优化步骤每个激发态与第一步第2个激发态的重叠积分,发现当前步骤的第1个态的重叠积分最大,因此当前步骤跟踪第1个态(Will follow Root 1)。结构优化的第3步则是将第3步的每个态和第2步的第1个态(而不是第1步的第2个态)作比较,选出重叠积分最大的态继续优化。
备注
(1)当重叠积分最大的2个态的重叠积分都很小,彼此差别不大时,程序会用激发能辅助态跟踪,即程序可能会跟踪重叠积分稍小、但激发能和前一步结构优化更接近的态。 (2)计算TDDFT数值Hessian也可进行态跟踪,当激发态存在近简并性时,这样做有助于避免在数值差分时跳到错误的激发态,导致数值差分结果不可靠。
基于sf-X2C/TDDFT-SOC的自旋轨道耦合计算
相对论效应包括标量相对论和自旋轨道耦合(spin-orbit coupling, SOC)。相对论计算需要使用 针对相对论效应优化的基组, 并选择合适的哈密顿 。BDF支持全电子的sf-X2C/TDDFT-SOC计算 [7, 12, 13] ,这里sf-X2C指用无自旋的精确二分量(eXact Two-Component, X2C)哈密顿考虑标量相对论效应,TDDFT-SOC指基于TDDFT计算自旋轨道耦合。注意虽然TDDFT是激发态方法,但TDDFT-SOC不仅可以用来计算SOC对激发态能量、性质的贡献,也可以用来计算SOC对基态能量、性质的贡献。
以基态为单重态的分子为例,完成sf-X2C/TDDFT-SOC计算需要按顺序调用三次TDDFT计算模块。其中,第一次执行利用R-TDDFT,计算单重态, 第二次利用SF-TDDFT计算三重态,最后一次读入前两个TDDFT计算的波函数,用态相互作用(State interaction, SI)方法 计算这些态的自旋轨道耦合。这从下面 \(\ce{CH2S}\) 分子的sf-X2C/TDDFT-SOC计算的高级输入可以清楚地看出。
$COMPASS
Title
ch2s
Basis # Notice: we use relativistic basis set contracted by DKH2
cc-pVDZ-DK
Geometry
C 0.000000 0.000000 -1.039839
S 0.000000 0.000000 0.593284
H 0.000000 0.932612 -1.626759
H 0.000000 -0.932612 -1.626759
End geometry
$END
$xuanyuan
heff # ask for sf-X2C Hamiltonian
3
hsoc # set SOC integral as 1e+mf-2e
2
$end
$scf
RKS
dft
PBE0
$end
#1st: R-TDDFT, calculate singlets
$tddft
isf
0
idiag
1
iroot
10
itda
0
istore # save TDDFT wave function in the 1st scratch file
1
$end
#2nd: spin-flip tddft, use close-shell determinant as reference to calculate triplets
$tddft
isf # notice here: ask for spin-flip up calculation
1
itda
0
idiag
1
iroot
10
istore # save TDDFT wave function in the 2nd scratch file, must be specified
2
$end
#3rd: tddft-soc calculation
$tddft
isoc
2
nprt # print level
10
nfiles
2
ifgs # whether to include the ground state in the SOC treatment. 0=no, 1=yes
1
imatsoc
8
0 0 0 2 1 1
0 0 0 2 2 1
0 0 0 2 3 1
0 0 0 2 4 1
1 1 1 2 1 1
1 1 1 2 2 1
1 1 1 2 3 1
1 1 1 2 4 1
imatrso
6
1 1
1 2
1 3
1 4
1 5
1 6
idiag # full diagonalization of SO Hamiltonian
2
$end
警告
计算必须按照isf=0,isf=1的顺序进行。当SOC处理不考虑基态(即
ifgs=0)时,计算的激发态数iroot越多,结果越准;当考虑基态(即ifgs=1)时,iroot太多反倒会令精度降低,具体表现为低估基态能量,此时iroot的选取没有固定规则,对于一般体系以几十为宜。若分子包含较重的元素(如第6、第7周期元素),BDF默认的栈内存往往不够大,需将OMP_STACKSIZE环境变量(当用intel编译器编译时,为KMP_STACKSIZE)设为较大的值,如4G或8G。
关键词 imatsoc 控制要打印哪些SOC矩阵元<A|hso|B>,
8表示要打印8组旋量态之间的SOC,下面顺序输入了8行整数数组;每一行的输入格式为
fileA symA stateA fileB symB stateB,代表矩阵元 <fileA,symA,stateA|hsoc|fileB,symB,stateB>,其中
fileA symA stateA代表文件fileA中的第symA个不可约表示的第stateA个根;例如1 1 1代表第1个TDDFT计算的第1个不可约表示的第1个根;
0 0 0表示基态
备注
程序每次最多只能打印4000个SOC矩阵元。
耦合矩阵元的打印输出如下,
[tddft_soc_matsoc]
Print selected matrix elements of [Hsoc]
SocPairNo. = 1 SOCmat = < 0 0 0 |Hso| 2 1 1 > Dim = 1 3
mi/mj ReHso(au) cm^-1 ImHso(au) cm^-1
0.0 -1.0 0.0000000000 0.0000000000 0.0000000000 0.0000000000
0.0 0.0 0.0000000000 0.0000000000 0.0000000000 0.0000000000
0.0 1.0 0.0000000000 0.0000000000 0.0000000000 0.0000000000
SocPairNo. = 2 SOCmat = < 0 0 0 |Hso| 2 2 1 > Dim = 1 3
mi/mj ReHso(au) cm^-1 ImHso(au) cm^-1
0.0 -1.0 0.0000000000 0.0000000000 0.0000000000 0.0000000000
0.0 0.0 0.0000000000 0.0000000000 0.0007155424 157.0434003237
0.0 1.0 0.0000000000 0.0000000000 -0.0000000000 -0.0000000000
SocPairNo. = 3 SOCmat = < 0 0 0 |Hso| 2 3 1 > Dim = 1 3
mi/mj ReHso(au) cm^-1 ImHso(au) cm^-1
0.0 -1.0 -0.0003065905 -67.2888361761 0.0000000000 0.0000000000
0.0 0.0 0.0000000000 0.0000000000 -0.0000000000 -0.0000000000
0.0 1.0 -0.0003065905 -67.2888361761 -0.0000000000 -0.0000000000
这里, < 0 0 0 |Hso| 2 2 1 > 表示矩阵元 <S0|Hso|T1> , 分别给出其实部ReHso和虚部ImHso。
由于S0只有一个分量,mi为0。T1(spin S=1)有3个分量(Ms=-1,0,1),用mj对这3个分量编号。
其中 Ms=0 的分量与基态的耦合矩阵元的虚部为 0.0007155424 au 。
警告
对比不同程序结果时需要注意:这里给出的是所谓spherical tensor,而不是cartesian tensor,即T1是T_{-1},T_{0},T_{1},不是Tx,Ty,Tz,两者之间存在酉变换。
SOC计算结果为,
Totol No. of States: 161 Print: 10
No. 1 w= -0.0006 eV
Spin: |Gs,1> 1-th Spatial: A1; OmegaSF= 0.0000eV Cr= 0.0000 Ci= 0.9999 Per:100.0%
SumPer: 100.0%
No. 2 w= 1.5481 eV
Spin: |S+,1> 1-th Spatial: A2; OmegaSF= 1.5485eV Cr= 0.9998 Ci= -0.0000 Per:100.0%
SumPer: 100.0%
No. 3 w= 1.5482 eV
Spin: |S+,3> 1-th Spatial: A2; OmegaSF= 1.5485eV Cr= 0.9998 Ci= 0.0000 Per:100.0%
SumPer: 100.0%
No. 4 w= 1.5486 eV
Spin: |S+,2> 1-th Spatial: A2; OmegaSF= 1.5485eV Cr= 0.9999 Ci= 0.0000 Per:100.0%
SumPer: 100.0%
No. 5 w= 2.2106 eV
Spin: |So,1> 1-th Spatial: A2; OmegaSF= 2.2117eV Cr= -0.9985 Ci= 0.0000 Per: 99.7%
SumPer: 99.7%
No. 6 w= 2.5233 eV
Spin: |S+,1> 1-th Spatial: A1; OmegaSF= 2.5232eV Cr= 0.9998 Ci= 0.0000 Per:100.0%
SumPer: 100.0%
No. 7 w= 2.5234 eV
Spin: |S+,3> 1-th Spatial: A1; OmegaSF= 2.5232eV Cr= 0.9998 Ci= -0.0000 Per:100.0%
SumPer: 100.0%
No. 8 w= 2.5240 eV
Spin: |S+,2> 1-th Spatial: A1; OmegaSF= 2.5232eV Cr= 0.0000 Ci= -0.9985 Per: 99.7%
SumPer: 99.7%
No. 9 w= 5.5113 eV
Spin: |S+,1> 1-th Spatial: B2; OmegaSF= 5.5115eV Cr= -0.7070 Ci= -0.0000 Per: 50.0%
Spin: |S+,3> 1-th Spatial: B2; OmegaSF= 5.5115eV Cr= 0.7070 Ci= 0.0000 Per: 50.0%
SumPer: 100.0%
No. 10 w= 5.5116 eV
Spin: |S+,1> 1-th Spatial: B2; OmegaSF= 5.5115eV Cr= -0.5011 Ci= -0.0063 Per: 25.1%
Spin: |S+,2> 1-th Spatial: B2; OmegaSF= 5.5115eV Cr= 0.7055 Ci= 0.0000 Per: 49.8%
Spin: |S+,3> 1-th Spatial: B2; OmegaSF= 5.5115eV Cr= -0.5011 Ci= -0.0063 Per: 25.1%
SumPer: 100.0%
*** List of SOC-SI results ***
No. ExEnergies Dominant Excitations Esf dE Eex(eV) (cm^-1)
1 -0.0006 eV 100.0% Spin: |Gs,1> 0-th A1 0.0000 -0.0006 0.0000 0.00
2 1.5481 eV 100.0% Spin: |S+,1> 1-th A2 1.5485 -0.0004 1.5487 12491.27
3 1.5482 eV 100.0% Spin: |S+,3> 1-th A2 1.5485 -0.0004 1.5487 12491.38
4 1.5486 eV 100.0% Spin: |S+,2> 1-th A2 1.5485 0.0001 1.5492 12494.98
5 2.2106 eV 99.7% Spin: |So,1> 1-th A2 2.2117 -0.0011 2.2112 17834.44
6 2.5233 eV 100.0% Spin: |S+,1> 1-th A1 2.5232 0.0002 2.5239 20356.82
7 2.5234 eV 100.0% Spin: |S+,3> 1-th A1 2.5232 0.0002 2.5239 20356.99
8 2.5240 eV 99.7% Spin: |S+,2> 1-th A1 2.5232 0.0008 2.5246 20362.08
9 5.5113 eV 50.0% Spin: |S+,1> 1-th B2 5.5115 -0.0002 5.5119 44456.48
10 5.5116 eV 49.8% Spin: |S+,2> 1-th B2 5.5115 0.0001 5.5122 44458.63
这里的输出有两部分,第一部分给出了每个 SOC-SI 态相对于S0态的能量及组成成分,例如
No. 10 w= 5.5116 eV表示第10个SOC-SI态的能量为5.5116 eV,注意这里是相对于S0态的能量;
下面三行是这个态的组成成分,
Spin: |S+,1> 1-th Spatial: B2;代表这是对称性为B2的第一个三重态(相对于S态自旋+1,因而是S+);
OmegaSF= 5.5115eV是相对于第一个旋量态的能量;
Cr= -0.5011 Ci= -0.0063是该成分在旋量态中组成波函数的实部与虚部,所占百分比为25.1%。
第二部分总结了SOC-SI态的计算结果,
ExEnergies列出考虑SOC后的激发能。Esf为原始不考虑SOC时的激发能;激发态表示用
Spin: |S,M> n-th sym来表示,自旋|Gs,1>,空间对称性为sym的第n个态。例如,|Gs,1>代表基态,|So,1>表示总自旋和基态相同的激发态,|S+,2>表示总自旋加1的激发态。M为自旋投影的第几个分量(in total 2S+1)。
关键词 imatrso 指定要计算并打印哪几组旋量态之间的跃迁偶极矩。这里指定打印 6 组跃迁偶极矩,
1 1表示基态固有偶极矩;
1 2表示第一个与第二个旋量态间的跃迁偶极矩。
备注
程序每次最多只能打印4000组跃迁偶极矩。
跃迁偶极矩的输出如下:
[tddft_soc_matrso]: Print selected matrix elements of [dpl]
No. ( I , J ) |rij|^2 E_J-E_I fosc rate(s^-1)
-------------------------------------------------------------------------------
1 1 1 0.472E+00 0.000000000 0.000000000 0.000E+00
Details of transition dipole moment with SOC (in a.u.):
<I|X|J> <I|Y|J> <I|Z|J> (also in debye)
Real= -0.113E-15 -0.828E-18 0.687E+00 -0.0000 -0.0000 1.7471
Imag= -0.203E-35 0.948E-35 0.737E-35 -0.0000 0.0000 0.0000
Norm= 0.113E-15 0.828E-18 0.687E+00
No. ( I , J ) |rij|^2 E_J-E_I fosc rate(s^-1)
-------------------------------------------------------------------------------
2 1 2 0.249E-05 1.548720567 0.000000095 0.985E+01
Details of transition dipole moment with SOC (in a.u.):
<I|X|J> <I|Y|J> <I|Z|J> (also in debye)
Real= -0.589E-03 0.207E-07 -0.177E-15 -0.0015 0.0000 -0.0000
Imag= -0.835E-08 0.147E-02 -0.198E-16 -0.0000 0.0037 -0.0000
Norm= 0.589E-03 0.147E-02 0.178E-15
提示
imatsoc设置为-1可指定打印所有的耦合矩阵元;默认不计算打印跃迁偶极矩,设置
imatrso为-1可以打印所有旋量态之间的跃迁偶极矩,设置imatrso为-2可以打印所有基态旋量态和所有激发态旋量态之间的跃迁偶极矩。SOC计算的参考态必须要么是RHF/RKS,要么是ROHF/ROKS,不支持UHF/UKS。
当SOC计算的参考态为ROHF/ROKS时,isf=0的TDDFT计算必须使用X-TDA(即itest=1, icorrect=1, isf=0, itda=1;不支持full X-TDDFT),isf=1的TDDFT计算必须使用SF-TDA(即isf=1, itda=1;不支持full SF-TDDFT)。
采用ECP基组的TDDFT-SOC自旋轨道耦合计算
除了sf-X2C全电子标量相对论哈密顿以外,也可以用赝势做TDDFT-SOC自旋轨道耦合计算,其中旋轨耦合赝势(SOECP)是首选,
为此需要选择合适的 旋轨耦合赝势基组 ,并在 xuanyuan 模块中设置 hsoc 为10(也可以写其它值,
但是都会当作10处理)。
其它输入与sf-X2C/TDDFT-SOC输入类似(例如在 scf 中指定轨道占据时要扣除芯层电子)或相同。
在下面的例子中,在 \(C_{2v}\) 点群对称性下计算了 InBr 分子的闭壳层基态 \(X^1\Sigma^+\) (A1)和最低三个激发态 \(^3\Pi\) (B1+B2)、 \(^1\Pi\) (B1+B2)、 \(^3\Sigma^+\) (A1),其中前两个Λ-S态是做了大量实验研究的束缚态, 后两个Λ-S态是排斥态,实验上不太关心。 输入中,首先在TDDFT级别下(这里采用Tamm-Dancoff近似)计算了Λ-S态的能量并存储波函,之后计算自旋轨道耦合后的Ω态能量。
$COMPASS
Title
soecp test: InBr
Basis-block
cc-pVTZ-PP
end basis
Geometry
In 0.0 0.0 0.0
Br 0.0 0.0 2.45
END geometry
group
C(2v) # Abelian symmetry must be used for SOC
$END
$XUANYUAN
hsoc
10
$END
$scf
rks
dft
pbe0
$end
$TDDFT
ISF
0
ITDA
1
istore
1
# 1Pi state: A1, A2, B1, B2
nroot
0 0 1 1
$END
$TDDFT
ISF
1
ITDA
1
istore
2
# 3Sigma+ and 3Pi states: A1, A2, B1, B2
nroot
1 0 1 1
$END
$TDDFT
isoc
2
nfiles
2
ifgs
1
idiag
2
$END
SOECP/TDDFT-SOC的计算输出与sf-X2C/TDDFT-SOC类似。结果总结如下,并与二分量EOM-CCSD的结果进行对比。
Λ-S态 |
TDDFT |
Ω态 |
TDDFT-SOC |
分裂 |
二分量EOM-CCSD |
分裂 |
|---|---|---|---|---|---|---|
\(X^1\Sigma^+\) |
0 |
0+ |
0 |
0 |
||
\(^3\Pi\) |
25731 |
0- |
24884 |
24516 |
||
0+ |
24959 |
75 |
24588 |
72 |
||
1 |
25718 |
759 |
25363 |
775 |
||
2 |
26666 |
948 |
26347 |
984 |
||
\(^1\Pi\) |
35400 |
1 |
35404 |
36389 |
||
\(^3\Sigma^+\) |
38251 |
0- |
38325 |
|||
1 |
38423 |
98 |
除了SOECP基组以外,也可以用标量ECP基组结合 有效核电荷近似(Zeff) 完成以上计算。 作为测试,首先删除Br基组中的SO赝势部分,重做上面的计算,但是会发现结果较差: \(^3\Pi_2\) 与 \(^3\Pi_1\) 的分裂只有850 cm \(^{-1}\) ,而 \(^3\Sigma^+\) 态的分裂几乎为零。 这是因为Br具有10个芯电子的ECP基组没有专门优化的有效核电荷,程序只能采用实际的核电荷数35:
SO-1e[BP]
Zeff for Wso
----------------------------------
IAtm ZA NCore Zeff
----------------------------------
1 49 28 SOECP
2 35 10 N.A.
----------------------------------
对于上例中的Br,不妨改用具有28个芯电子的标量ECP基组cc-pVTZ-ccECP,基组的输入部分修改如下:
Basis-block
cc-pvtz-pp
Br=cc-pvtz-ccecp
end basis
在 xuanyuan 之后的模块中未指定轨道占据,因此无需修改输入。在TDDFT-SOC计算输出的一开始可以看到
SO-1e[BP]
Zeff for Wso
----------------------------------
IAtm ZA NCore Zeff
----------------------------------
1 49 28 SOECP
2 35 28 1435.000
----------------------------------
这表明在Br的单电子自旋轨道积分中,用优化好的1435.000替换默认的核电荷数35(一般来说,ECP芯电子数NCore越大,有效核电荷Zeff越大), 而对In原子仍旧计算SOECP积分。计算结果如下,可见旋轨分裂得到了明显改善:
Λ-S态 |
TDDFT |
Ω态 |
Br:SOECP |
分裂 |
Br:ECP |
分裂 |
|---|---|---|---|---|---|---|
\(X^1\Sigma^+\) |
0 |
0+ |
0 |
0 |
||
\(^3\Pi\) |
25731 |
0- |
24884 |
25019 |
||
0+ |
24959 |
75 |
25084 |
65 |
||
1 |
25718 |
759 |
25856 |
772 |
||
2 |
26666 |
948 |
26808 |
952 |
||
\(^1\Pi\) |
35400 |
1 |
35404 |
35729 |
||
\(^3\Sigma^+\) |
38251 |
0- |
38325 |
38788 |
||
1 |
38423 |
98 |
38853 |
65 |
最后,TDDFT-SOC计算也可以用SOECP(或标量ECP)基组与全电子非相对论基组进行组合。 BDF程序已经对Xe之前主族元素的全电子非相对论基组优化了Zeff(较重的稀有气体元素除外)。 例如,In继续用cc-pVTZ-PP,而Br用全电子非相对论基组cc-pVTZ,会得到与SOECP/TDDFT-SOC相近的结果。详细结果从略。
注意
用有效核电荷方法进行TDDFT-SOC计算时的注意事项:必须用 优化好的有效核电荷 才能保证精度。为此要检查输出文件打印的Zeff值,尽量不要出现N.A.,这对ECP基组尤其重要。
SOECP或标量ECP与全电子基组组合时,关于全电子基组的注意事项:由于使用全电子基组的原子不考虑标量相对论相应,因此不能是重原子,且必须用非相对论基组。
一阶非绝热耦合矩阵元(fo-NACME)的计算
如前所述,(一阶)非绝热耦合矩阵元 [40, 41, 42] 在非辐射跃迁过程中有着重要的意义,其主要用途之一为计算内转换速率常数(参见 BDF-MOMAP联用计算内转换速率常数的示例 )。在BDF中,基态和激发态之间的NACME,以及激发态和激发态之间的NACME的输入文件在写法上存在一定差异,以下分别介绍。
备注
基态和激发态之间的NACME、激发态和激发态之间的NACME均支持R-TDDFT、U-TDDFT、R-TDA和U-TDA。自2026年1月起,也支持X-TDDFT和X-TDA。
(1)基态和激发态之间的NACME: \(\ce{NO3}\) 自由基的D0/D1 NACME(GB3LYP/cc-pVDZ)
$COMPASS
Title
NO3 radical NAC, 1st excited state
Basis
cc-pvdz
Geometry
N 0.0000000000 0.0000000000 -0.1945736441
O -2.0700698389 0.0000000000 -1.1615808530
O 2.0700698389 -0.0000000000 -1.1615808530
O -0.0000000000 0.0000000000 2.4934136445
End geometry
unit
bohr
$END
$XUANYUAN
$END
$SCF
UKS
dft
GB3LYP
spinmulti
2
$END
$tddft
iroot
1 # One root for each irrep
istore
1 # File number, to be used later in $resp
crit_vec
1.d-6
crit_e
1.d-8
gridtol
1.d-7 # tighten the tolerance value of XC grid generation. This helps to
# reduce numerical error, and is recommended for open-shell molecules
$end
$resp
iprt
1
QUAD # quadratic response
FNAC # first-order NACME
single # calculation of properties from single residues (ground state-excited
# state fo-NACMEs belong to this kind of properties)
norder
1
method
2
nfiles
1 # must be the same as the istore value in the $TDDFT block
states
1 # Number of excited states for which NAC is requested.
# First number 1: read TDDFT results from file No. 1
# Second number 2: the second irrep, in this case A2
# (note: this is the pair symmetry of the particle-hole pair, not
# the excited state symmetry. One must bear this in mind because the
# ground state of radicals frequently does not belong to the totally
# symmetric irrep)
# If no symmetry is used, simply use 1.
# Third number 1: the 1st excited state that belongs to this irrep
1 2 1
$end
注意 $resp 模块中指定的不可约表示为pair irrep(即跃迁涉及的占据轨道和空轨道的不可约表示的直积;对于阿贝尔点群,pair irrep可以由基态不可约表示和激发态不可约表示的直积求得),而不是激发态的irrep。该分子的基态(D0)属于B1不可约表示,第一二重态激发态(D1)属于B2不可约表示,因此D1态的pair irrep为B1和B2的直积,即A2。Pair irrep也可由TDDFT模块的输出读取得到,即以下输出部分的Pair一栏:
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 A2 1 B2 0.8005 eV 1548.84 nm 0.0000 0.0186 98.2% CO(bb): B2( 2 )-> B1( 5 ) 3.992 0.622 0.0000
2 B1 1 A1 1.9700 eV 629.35 nm 0.0011 0.0399 92.2% CO(bb): A1( 8 )-> B1( 5 ) 3.958 0.667 1.1695
3 B2 1 A2 2.5146 eV 493.06 nm 0.0000 0.0384 98.4% CO(bb): A2( 1 )-> B1( 5 ) 4.159 0.319 1.7141
4 A1 2 B1 2.6054 eV 475.87 nm 0.0171 0.0154 87.7% CO(bb): B1( 4 )-> B1( 5 ) 3.984 0.746 1.8049
计算完成后,在 $resp 模块的输出部分的结尾,可以看到NACME的计算结果:
Gradient contribution from Final-NAC(R)-Escaled
1 0.0000000000 -0.0000000000 0.0000000000
2 -0.0000000000 -0.1902838724 0.0000000000
3 -0.0000000000 0.1902838724 0.0000000000
4 -0.0000000000 0.0000000000 0.0000000000
可以发现计算结果有N行(其中N为体系的原子数),每行有3个实数,分别代表该原子的NACME的x、y、z分量。注意该结果没有包括电子平移因子(electron translation factor, ETF)的贡献,对于某些分子,不包括ETF的NACME可能会不具有平移不变性,进而导致后续动力学模拟等计算产生误差。此时需要使用考虑了ETF的NACME,在输出文件稍后的位置可以读取得到:
Gradient contribution from Final-NAC(S)-Escaled
1 0.0000000000 -0.0000000000 0.0000000000
2 -0.0000000000 -0.1920053581 0.0000000000
3 -0.0000000000 0.1920053581 0.0000000000
4 -0.0000000000 0.0000000000 -0.0000000000
程序还会输出名为dpq-R、Final-NAC(R)、dpq-S、Final-NAC(S)等的矢量,这些量是中间变量,仅供监测计算过程使用,并非最终的NACME,一般情况下用户可忽略这些输出。
(2)激发态和激发态之间的NACME:苯乙酮的T1/T2 NACME(BH&HLYP/def2-SVP)
$compass
title
PhCOMe
basis
def2-SVP
geometry
C -0.3657620861 4.8928163606 0.0000770328
C -2.4915224786 3.3493223987 -0.0001063823
C -2.2618953860 0.7463412225 -0.0001958732
C 0.1436118499 -0.3999193588 -0.0000964543
C 2.2879147462 1.1871091769 0.0000824391
C 2.0183382809 3.7824607425 0.0001740921
H -0.5627800515 6.9313968857 0.0001389666
H -4.3630645857 4.1868310874 -0.0002094148
H -3.9523568496 -0.4075513123 -0.0003833263
H 4.1604797959 0.3598389310 0.0001836001
H 3.6948496439 4.9629708946 0.0003304312
C 0.3897478526 -3.0915327760 -0.0002927344
O 2.5733215239 -4.1533492423 -0.0002053903
C -1.8017552120 -4.9131221777 0.0003595831
H -2.9771560760 -4.6352720097 1.6803279168
H -2.9780678476 -4.6353463569 -1.6789597597
H -1.1205416224 -6.8569277129 0.0002044899
end geometry
unit
bohr
nosymm
$end
$XUANYUAN
$END
$SCF
rks
dft
bhhlyp
$END
$tddft
isf # request for triplets (spin flip up)
1
ialda # use collinear kernel (NAC only supports collinear kernel)
4
iroot
2 # calculate T1 and T2 states
crit_vec
1.d-6
crit_e
1.d-8
istore
1
iprt
2
$end
$resp
iprt
1
QUAD
FNAC
double # calculation of properties from double residues (excited state-excited
# state fo-NACMEs belong to this kind of properties)
norder
1
method
2
nfiles
1
pairs
1 # Number of pairs of excited states for which NAC is requested.
1 1 1 1 1 2
noresp # do not include the quadratic response contributions (recommended)
$end
计算得到T1态和T2态的NACME:
Gradient contribution from Final-NAC(R)-Escaled
1 0.0005655253 0.0005095355 -0.2407937116
2 -0.0006501682 -0.0005568029 0.5339003311
3 0.0009640605 0.0003767996 -2.6530192038
4 -0.0013429266 -0.0034063171 1.6760344312
5 0.0010446538 0.0006384285 -0.8024123329
6 -0.0001081722 -0.0006245719 -0.0487310115
7 -0.0000001499 0.0000176176 -0.0730900968
8 -0.0000214634 0.0000165092 0.3841606239
9 0.0000026057 -0.0000025322 -0.2553378323
10 -0.0002028358 -0.0000591642 0.5800987974
11 -0.0000166820 0.0000105734 0.2713836450
12 -0.0023404123 0.0052038311 3.5121827769
13 0.0021749503 -0.0012164868 -2.7480141157
14 0.0000433873 -0.0011202812 0.2896243729
15 0.1407516324 0.1432264573 -0.1655701318
16 -0.1407399684 -0.1429881941 -0.1657943551
17 -0.0000034197 0.0004577563 -0.0833951446
激发态的定域化
对于有机分子聚集体等体系,总体系的激发态往往是离域在整个体系上的。通过激发态定域化,可以把总体系的最低几个激发态进行线性组合,得到并非能量本征态、但基本局域在单个分子上的透热态,同时得到各个透热态之间的耦合矩阵元。这一操作对于计算分子间能量转移速率(例如用Marcus理论、非绝热动力学、MCTDH、TD-DMRG等方法)、分析激发态成分等有重要意义。
以下例子计算了两个乙烯分子组成的体系的最低4个激发态,并对其进行定域化 [73] :
$COMPASS
Basis
cc-pvdz
Geometry
C 0.000000 0.000000 0.000000
C 1.332000 0.000000 0.000000
H -0.574301 -0.928785 0.000000
H -0.574301 0.928785 0.000000
H 1.906301 0.928785 0.000000
H 1.906301 -0.928785 0.000000
C -0.000000 0.000000 3.5000
C 1.332000 -0.000000 3.5000
H -0.574301 0.928785 3.50000
H -0.574301 -0.928785 3.50000
H 1.906301 -0.928785 3.50000
H 1.906301 0.928785 3.50000
End geometry
Group
C(1)
Nfragment # must input: number of fragment, should be 1
1
$END
$xuanyuan
$end
$scf
rks
dft
B3lyp
$end
$TDDFT
ITDA
1
IDIAG
1
istore
1
iroot
4
crit_e # set a small threshhold for TDDFT energy convergence
1.d-8
$END
# calculate local excited states (LOCALES)
$elecoup
locales
1
$END
&database
fragment 1 12 # first fragment with 12 atoms, next line gives the atom list
1 2 3 4 5 6 7 8 9 10 11 12
&end
TDA计算了4个激发态,输出如下,
No. Pair ExSym ExEnergies Wavelengths f D<S^2> Dominant Excitations IPA Ova En-E1
1 A 2 A 7.4870 eV 165.60 nm 0.0000 0.0000 82.6% CV(0): A( 16 )-> A( 17 ) 13.476 0.820 0.0000
2 A 3 A 8.6807 eV 142.83 nm 0.0673 0.0000 79.6% CV(0): A( 16 )-> A( 18 ) 14.553 0.375 1.1937
3 A 4 A 9.0292 eV 137.31 nm 0.0000 0.0000 62.4% CV(0): A( 16 )-> A( 20 ) 15.353 0.398 1.5422
4 A 5 A 9.0663 eV 136.75 nm 0.0000 0.0000 50.4% CV(0): A( 15 )-> A( 18 ) 15.688 0.390 1.5793
定域化的过程及定域的激发态为,
No. 8 iteration
Pair States : 1 2
aij,bij,abij -.25659893E-01 -.48045653E-11 0.25659893E-01
cos4a,sin4a 0.10000000E+01 -.18724027E-09
cosa,sina 0.10000000E+01 0.00000000E+00
Pair States : 1 3
aij,bij,abij -.40325646E-02 0.35638586E-11 0.40325646E-02
cos4a,sin4a 0.10000000E+01 0.88376974E-09
cosa,sina 0.10000000E+01 0.00000000E+00
Pair States : 1 4
aij,bij,abij -.25679319E-01 -.28753641E-08 0.25679319E-01
cos4a,sin4a 0.10000000E+01 -.11197197E-06
cosa,sina 0.10000000E+01 0.27877520E-07
Pair States : 2 3
aij,bij,abij -.39851115E-02 -.27118892E-05 0.39851124E-02
cos4a,sin4a 0.99999977E+00 -.68050506E-03
cosa,sina 0.99999999E+00 0.17012628E-03
Pair States : 2 4
aij,bij,abij -.42686102E-02 -.95914926E-06 0.42686103E-02
cos4a,sin4a 0.99999997E+00 -.22469825E-03
cosa,sina 0.10000000E+01 0.56174562E-04
Pair States : 3 4
aij,bij,abij -.67873307E-01 -.47952471E-02 0.68042488E-01
cos4a,sin4a 0.99751360E+00 -.70474305E-01
cosa,sina 0.99984454E+00 0.17632279E-01
Sum= 0.13608498 Max Delta= 0.00531009
No. 9 iteration
Pair States : 1 2
aij,bij,abij -.40325638E-02 0.35621782E-11 0.40325638E-02
cos4a,sin4a 0.10000000E+01 0.88335323E-09
cosa,sina 0.10000000E+01 0.00000000E+00
Pair States : 1 3
aij,bij,abij -.25690755E-01 -.11200070E-08 0.25690755E-01
cos4a,sin4a 0.10000000E+01 -.43595721E-07
cosa,sina 0.10000000E+01 0.10536712E-07
Pair States : 1 4
aij,bij,abij -.25690755E-01 -.10900573E-11 0.25690755E-01
cos4a,sin4a 0.10000000E+01 -.42429944E-10
cosa,sina 0.10000000E+01 0.00000000E+00
Pair States : 2 3
aij,bij,abij -.41480079E-02 -.83549288E-06 0.41480080E-02
cos4a,sin4a 0.99999998E+00 -.20142027E-03
cosa,sina 0.10000000E+01 0.50355067E-04
Pair States : 2 4
aij,bij,abij -.41480100E-02 0.17462423E-06 0.41480100E-02
cos4a,sin4a 0.10000000E+01 0.42098314E-04
cosa,sina 0.10000000E+01 0.10524580E-04
Pair States : 3 4
aij,bij,abij -.68042492E-01 0.19389042E-08 0.68042492E-01
cos4a,sin4a 0.10000000E+01 0.28495490E-07
cosa,sina 0.10000000E+01 0.74505806E-08
Sum= 0.13608498 Max Delta= 0.00000000
***************** Diabatic Hamiltonian matrix ****************
State1 State2 State3 State4
State1 7.486977 0.000000 0.000000 0.000000
State2 0.000000 9.029214 -0.000020 0.000021
State3 0.000000 -0.000020 8.873501 0.192803
State4 0.000000 0.000021 0.192803 8.873501**************************************************************
在Diabatic Hamiltonian matrix中,对角元为定域激发态的能量,非对角元为两个定域态之间的耦合,这里的能量单位是 eV 。耦合矩阵元可以代入Marcus理论等的公式,得到能量转移或电子转移的速率。例如,若读取的是LE态和CT态之间的耦合矩阵元,则可用来计算分子在已经激发到LE态的情况下,发生电子转移(或者说电荷分离)的速率;若读取的是两个LE态之间的耦合矩阵元,但这两个LE态局域在不同的分子上,则可用来计算这两个分子之间的能量转移速率。